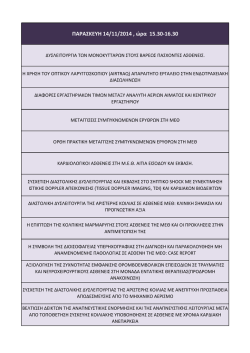
ΔΦΜΕ-E.4200-4/2 ΣΥΝΟΔΕΥΤΙΚΗ ΕΠΙΣΤΟΛΗ ΑΙΤΗΣΗΣ ΚΛΙΝΙΚΗΣ ΔΟΚΙΜΗΣ ΣΤΟΝ ΕΟΦ Α. ΒΑΣΙΚΕΣ ΠΛΗΡΟΦΟΡΙΕΣ ΤΗΣ ΚΛΙΝΙΚΗΣ ΔΟΚΙΜΗΣ 1. Αριθμός EudraCT: Φάση: I II III IV 2. Κωδικός αριθμός πρωτοκόλλου: 3. Τίτλος πρωτοκόλλου: 4. Χορηγός: Εμπορικός Μη Εμπορικός 5. Αιτών: 6. Εξουσιοδοτημένο CRO στην Ελλάδα: 7. Στοιχεία επιτηρητή της μελέτης (ονοματεπώνυμο και εταιρεία): 8. Αριθμός ερευνητικών κέντρων στην Ελλάδα: 9. Προβλεπόμενος συνολικός αριθμός ασθενών στην Ελλάδα: 10. Χρονική διάρκεια της κλινικής δοκιμής στην Ελλάδα: .....Έτη .....Μήνες ....Ημέρες 11. Συνολικός προϋπολογισμός της κλινικής δοκιμής (€): 12. Κατά την ημ/νία υποβολής της αίτησης κλινικής δοκιμής στον ΕΟΦ έχει επίσης υποβληθεί αντίστοιχη αίτηση στις αρμόδιες αρχές των ακόλουθων χωρών της Ε.Ε.: ΒΕ NL ΒG ΑΤ CZ PL DK PT DΕ RO EE SI IE SK ES FI FR SE IT UK CY LV LT LU HU MT 13. Κατά την ημ/νία υποβολής της αίτησης κλινικής δοκιμής στον ΕΟΦ έχει χορηγηθεί άδεια διεξαγωγής της κλινικής δοκιμής από τις αρμόδιες αρχές των ακόλουθων χωρών της Ε.Ε.: ΒΕ NL ΒG ΑΤ CZ PL DK PT DΕ RO EE SI IE SK ES FI FR SE IT UK CY LV LT LU 14. Στην κλινική δοκιμή προβλέπεται η ύπαρξη ανεξάρτητης επιτροπής παρακολούθησης ασφάλειας/δεδομένων (π.χ. IDMC, DSMB, DMC); 15. Η αίτηση συνοδεύεται από επιστημονική συμβουλή του ΕΜΑ για την κλινική δοκιμή; HU MT ΟΧΙ ΟΧΙ ΝΑΙ ΝΑΙ 16. Η κλινική δοκιμή αποτελεί ή πρόκειται να αποτελέσει τμήμα ενός σχεδίου παιδιατρικής έρευνας (PIP); 17. Η κλινική δοκιμή περιλαμβάνει κάποια υπομελέτη; ΟΧΙ ΟΧΙ ΝΑΙ ΝΑΙ 18. Εφόσον η κλινική δοκιμή περιλαμβάνει φαρμακογενετική μελέτη, συνυποβάλλεται υπεύθυνη δήλωση του χορηγού ότι θα καταστρέψει άμεσα τυχόν δείγματα της φαρμακογενετικής μελέτης μόλις αυτό ζητηθεί από την αρμόδια αρχή; ΟΧΙ ΝΑΙ 19. Εφόσον πρόκειται για επανυποβολή, επισημαίνονται οι αλλαγές σε σχέση με την προηγούμενη υποβολή; ΟΧΙ ΝΑΙ Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 1 από 25 ΔΦΜΕ-E.4200-4/2 Β. ΠΛΗΡΟΦΟΡΙΕΣ ΓΙΑ ΤΑ ΥΠΟ ΕΡΕΥΝΑ ΦΑΡΜΑΚΕΥΤΙΚΑ ΠΡΟΪΟΝΤΑ (IMP)1 1. Οι Πληροφορίες Ασφαλείας Αναφοράς (RSI) βρίσκονται στο: 2. Πρόκειται για την πρώτη χορήγηση σε ανθρώπους; 3. Το IMP είναι ναρκωτική και ψυχοτρόπος ουσία; ΙΒ SPC ΟΧΙ Ενότητα: .......... ΝΑΙ ΟΧΙ ΝΑΙ 4. Ποσότητα που πρόκειται να χρησιμοποιηθεί (περιγράφονται αναλυτικά οι ποσότητες για όλες τις φαρμακοτεχνικές μορφές και όλες τις περιεκτικότητες του IMP): Γ. ΠΛΗΡΟΦΟΡΙΕΣ ΓΙΑ ΤΑ ΜΗ ΥΠΟ ΕΡΕΥΝΑ ΦΑΡΜΑΚΕΥΤΙΚΑ ΠΡΟΪΟΝΤΑ (NIMP)2 1. Στην κλινική δοκιμή γίνεται χρήση κάποιου NIMP; ΟΧΙ ΝΑΙ3 - Εάν ΝΑΙ, αναφέρετε το όνομα της δραστικής ουσίας: - Εάν ΝΑΙ, αναφέρετε το εμπορικό όνομα του NIMP (εφόσον δεν ισχύει η περίπτωση 2 κάτωθι): 2. Στην κλινική δοκιμή δεν θα χρησιμοποιηθεί NIMP με συγκεκριμένο εμπορικό όνομα αλλά οποιοδήποτε αδειοδοτημένο στην Ελλάδα NIMP με την ανωτέρω δραστική ουσία; ΟΧΙ ΝΑΙ 3. Κατηγορία στην οποία ανήκει το NIMP4: Ι. Rescue medication ΙΙ. Challenge agent ΙΙΙ. Medicinal products used to assess end-points in the clinical trial IV. Concomitant medicinal products systematically prescribed to the study patients V. Background treatment 4. Το NIMP έχει άδεια κυκλοφορίας στην Ελλάδα; 5. Το NIMP έχει άδεια κυκλοφορίας σε άλλη χώρα της Ε.Ε; 6. Το ΝIMP είναι ναρκωτική και ψυχοτρόπος ουσία; ΟΧΙ ΟΧΙ ΝΑΙ ΝΑΙ ΟΧΙ ΝΑΙ Δ. ΛΟΙΠΕΣ ΠΛΗΡΟΦΟΡΙΕΣ 1. Αναφέρετε οποιεσδήποτε άλλες πληροφορίες θεωρείτε σκόπιμο ότι πρέπει να συμπεριληφθούν στη συνοδευτική επιστολή π.χ. ιδιομορφίες της κλινικής δοκιμής, ειδικά χαρακτηριστικά των συμμετεχόντων (π.χ. ανήλικοι, ανίκανοι για συγκατάθεση, κ.ά): Υπογραφή & σφραγίδα χορηγού Ημ/νία: / /201 1 Στην περίπτωση ύπαρξης και άλλων IMP επαναλάβετε την ενότητα Β αντιστοίχως για κάθε IMP. Δεν χρειάζεται επανάληψη για IMP διαφορετικής περιεκτικότητας ή μορφής. 2 Στην περίπτωση ύπαρξης και άλλων NIMP επαναλάβετε την ενότητα Γ αντιστοίχως για κάθε NIMP. Δεν χρειάζεται επανάληψη για IMP διαφορετικής περιεκτικότητας ή μορφής. 3 Εάν ΝΑΙ, βεβαιωθείτε ότι η αίτηση συνοδεύται από την κατάλληλη τεκμηρίωση σύμφωνα με το Annex 2 του Guidance On Investigational Medicinal Products (IMPs) And 'Non Investigational Medicinal Products' (NIMPS) (Rev. 1, March 2011) 4 Σύμφωνα με το Annex 1 του Guidance On Investigational Medicinal Products (IMPs) And 'Non Investigational Medicinal Products' (NIMPS) (Rev. 1, March 2011) Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 2 από 25 ΔΦΜΕ-E.4200-4/2 ΛΙΣΤΑ ΕΛΕΓΧΟΥ ΤΩΝ ΥΠΟΒΑΛΛΟΜΕΝΩΝ ΔΙΚΑΙΟΛΟΓΗΤΙΚΩΝ (βάσει των σχετικών ενοτήτων της εγκυκλίου ΕΟΦ Α.Π. 88148/23.12.2010 καθώς και της εγκυκλίου ΕΟΦ Α.Π. 22744/20.3.2013 ως προς τον τρόπο υποβολής) 1 Το σύνολο του υποβαλλόμενου φακέλου και το σχετικό XML περιλαμβάνεται στο CD-ROM ΝΑΙ ΟΧΙ 2 Συνοδευτική επιστολή (ΔΦΜΕ-Ε.4200-2) με τα περιεχόμενα που ορίζονται στην ενότητα 2.3 ΝΑΙ ΟΧΙ 3 Επιβεβαίωση παραλαβής αριθμού EudraCT ΝΑΙ ΟΧΙ 4 Έντυπο αίτησης (CTA) στα Ελληνικά (ΔΦΜΕ-Ε.4200-1) ΝΑΙ ΟΧΙ 5 Eπιστολή εξουσιοδότησης στον αιτούντα να ενεργεί εξ ονόματος του χορηγού, εάν ο αιτών δεν είναι ο χορηγός ΝΑΙ ΟΧΙ 6 Eπιστολή εξουσιοδότησης στο νόμιμο εκπρόσωπο του χορηγού στην Ε.Ε. εάν είναι διαφορετικός από το χορηγό ΝΑΙ ΟΧΙ 7 Κατάλογος Αρμοδίων Αρχών προς τις οποίες έχει υποβληθεί η αίτηση μαζί με τη σχετική απόφαση, σε περίπτωση που έχει εκδοθεί ΝΑΙ ΟΧΙ 8 Παράβολο με αναφορά σε στοιχεία ταυτοποίησης της κλινικής δοκιμής ΝΑΙ ΟΧΙ 9 Περίληψη του πρωτοκόλλου (στην Ελληνική) ΝΑΙ ΟΧΙ 10 Πρωτόκολλο με τα περιεχόμενα που ορίζονται στην ενότητα 2.5 (στην Ελληνική) ΝΑΙ ΟΧΙ 11 Υπογεγραμμένο συμφωνητικό μεταξύ χορηγού και ερευνητή ΝΑΙ ΟΧΙ 12 Σύντομο και πρόσφατο βιογραφικό σημείωμα των κύριων ερευνητών για όλα τα κέντρα στα οποία διεξάγεται η μελέτη ΝΑΙ ΟΧΙ 13 Έντυπο ενημέρωσης και συγκατάθεσης των συμμετεχόντων (κωδικός, έκδοση, ημερομηνία) ΝΑΙ ΟΧΙ 14 IB ή το έγγραφο που αντικαθιστά το IB, όπως ορίζεται στην ενότητα 2.6 ΝΑΙ ΟΧΙ 15 IMPD/απλοποιημένος IMPD, όπως ορίζεται στις ενότητες 2.7 και 2.7.3 ΝΑΙ ΟΧΙ 16 Συμμόρφωση με τους Κανόνες Καλής Παραγωγής (GMP) για τα IMPs, όπως ορίζονται στην ενότητα 2.7.1. ΝΑΙ ΟΧΙ 17 Δεδομένα για την ποιότητα των IMPs, όπως ορίζονται στην ενότητα 2.7.2.2 ΝΑΙ ΟΧΙ 18 Φάκελος NIMP, όπως ορίζεται στην ενότητα 2.8 καθώς και στο Annex 2 του “Guidance On Investigational Medicinal Products (IMPs) and Non Investigational Medicinal Products (NIMPS) (Rev. 1, March 2011)” ΝΑΙ ΟΧΙ 19 Τα συμπληρωματικά τμήματα της τεκμηρίωσης, όπως ορίζονται στην ενότητα 2.9 ΝΑΙ ΟΧΙ Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 3 από 25 ΔΦΜΕ-E.4200-4/2 Παράρτημα 1: Έντυπο Αίτησης ΑΙΤΗΣΗ ΓΙΑ ΧΟΡΗΓΗΣΗ ΑΔΕΙΑΣ ΔΙΕΞΑΓΩΓΗΣ ΚΛΙΝΙΚΗΣ ΔΟΚΙΜΗΣ ΦΑΡΜΑΚΕΥΤΙΚΟΥ ΠΡΟΙΟΝΤΟΣ ΑΝΘΡΩΠΙΝΗΣ ΧΡΗΣΗΣ ΠΡΟΣ ΤΟΝ ΕΟΦ ΚΑΙ ΓΙΑ ΓΝΩΜΟΔΟΤΗΣΗ ΤΗΣ ΕΘΝΙΚΗΣ ΕΠΙΤΡΟΠΗΣ ΔΕΟΝΤΟΛΟΓΙΑΣ Συμπληρώνεται από την υπηρεσία: Ημερομηνία παραλαβής της αίτησης: Ημερομηνία απαίτησης επιπλέον στοιχείων (κατά την επικύρωση) ώστε να καταστεί έγκυρη η αίτηση: Ημερομηνία έγκυρης αίτησης: Ημερομηνία αίτησης πρόσθετων πληροφοριών (κατά την αξιολόγηση): Λόγοι μη αποδοχής/ αρνητικής γνωμοδότησης: Ημερομηνία: Ημερομηνία παραλαβής πρόσθετων / τροποποιημένων πληροφοριών: Ημερομηνία έναρξης διαδικασίας: Χορήγηση αδείας / θετική γνωμοδότηση: Ημερομηνία: Κωδικός αριθμός ΕΟΦ: Κωδικός αριθμός ΕΕΔ: Ανάκληση της αίτησης: Ημερομηνία: Συμπληρώνεται από τον αιτούντα: Οι ερωτήσεις στο παρόν έντυπο για αίτηση χορήγησης αδείας από τον ΕΟΦ έχουν επίσης σχέση με τη γνωμοδότηση από την ΕΕΔ (αντιπροσωπεύει το τμήμα 1 του εντύπου της αίτησης προς την ΕΕΔ) και μπορούν να χρησιμοποιηθούν ως τμήμα της αίτησης προς την ΕΕΔ.Υποδείξτε το σκοπό της αίτησης σημειώνοντας σε ένα από τα παρακάτω τετραγωνίδια: ΑΙΤΗΣΗ ΠΡΟΣ ΤΟΝ ΕΟΦ ΓΙΑ ΧΟΡΗΓΗΣΗ ΑΔΕΙΑΣ: ΑΙΤΗΣΗ ΓΙΑ ΓΝΩΜΟΔΟΤΗΣΗ ΤΗΣ ΕΘΝΙΚΗΣ ΕΠΙΤΡΟΠΗΣ ΔΕΟΝΤΟΛΟΓΙΑΣ: Α. ΣΤΟΙΧΕΙΑ ΚΛΙΝΙΚΗΣ ΔΟΚΙΜΗΣ Α.1 Α.2 Α.3 A.3.1 A 3.2 Α.4 Α.5 Α.6 Α.7 A. 8 Κράτος μέλος στο οποίο υποβάλλεται η αίτηση: Αριθμός EudraCT Πλήρης τίτλος της δοκιμής: Τίτλος δοκιμής για μη ειδικούς, σε κατανοητή ,π.χ. μη ειδική φρασεολογία: Ονομασία ή συντομογραφία του τίτλου της δοκιμής εάν υπάρχει: Κωδικός αριθμός, έκδοση και ημερομηνία πρωτοκόλλου του χορηγού5: Επιπλέον διεθνή αναγνωριστικά στοιχεία δοκιμής(π.χ. WHO, ISRCTN,6 Αριθμός US NCT7) αν είναι διαθέσιμα Αποτελεί η παρούσα αίτηση επανυποβολή ναι όχι Εάν ναι, υποδείξτε το γράμμα επανυποβολής8 Είναι η δοκιμή μέρος ενός Σχεδίου Παιδιατρικής Έρευνας (ΡΙΡ) ναι όχι Αριθμός Απόφασης EMEA για Σχέδιο Παιδιατρικής Έρευνας (ΡΙΡ) 5 Κάθε μετάφραση του πρωτοκόλλου θα πρέπει να φέρει την ίδια ημερομηνία και αριθμό έκδοσης όπως εκείνης του πρωτοτύπου κειμένου 2 Αριθμός Διεθνούς Προτύπου Τυχαιοποιημένων Ελεγχόμενων Δοκιμών(ISRCTN). Οι χορηγοί μπορεί να επιθυμούν τη χρήση ISRCTN για να ταυτοποιήσουν την δοκιμή τους επί πλέον του αριθμού EudraCT π.χ. εάν η δοκιμή τους είναι μέρος μιας πολυεθνικής δοκιμής με κέντρα εκτός της Κοινότητας. Οι χορηγοί μπορούν να αποκτήσουν τον αριθμό και καθοδήγηση από την ιστοσελίδα των ελεγχόμενων δοκιμών που βρίσκονται σε εξέλιξη http://www.contolled-trials.com/isrctn για τον οποίο υπάρχει υπέρ-σύνδεσμος από την βάση δεδομένων του EudraCT http://eudract.emea.europe.eu/. Όποτε είναι διαθέσιμος ο παραπάνω αριθμός , οι χορηγοί θα πρέπει να τον δηλώνουν στο τμήμα Α.6 του εντύπου της αίτησης. 3 Οι αριθμοί US National Clinical Trial (NCT) απαιτούνται στην αίτηση για κλινικές δοκιμές του FDA. 4 Προκειμένου για επανυποβολή μετά από προηγούμενη ανάκληση μιας αίτησης ή αρνητική γνωμοδότηση της ΕΕΔ, ή προηγούμενη ανάκληση μιας αίτησης ή αρνητικής απάντησης από τον ΕΟΦ, προσθέστε ένα γράμμα που αυξάνει με κάθε επανυποβολή Α για πρώτη επανυποβολή, Β για δεύτερη, Γ για τρίτη και ούτω καθ’ εξής. Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 4 από 25 ΔΦΜΕ-E.4200-4/2 Β. ΣΤΟΙΧΕΙΑ XOΡΗΓΟΥ ΥΠΕΥΘΥΝΟΥ ΓΙΑ ΤΗΝ ΑΙΤΗΣΗ Β.1 ΧΟΡΗΓΟΣ Β.1.1 Επωνυμία οργανισμού: Β.1.2 Ονοματεπώνυμο υπεύθυνου επικοινωνίας: Β1.2.1 Όνομα Β1.2.2 Μεσαίο Β1.2.3 Επίθετο Β 1.3 Διεύθυνση: Β1.3.1 Οδός Β1.3.2 Πόλη Β1.3.3 Ταχυδρομικός Κώδικας Β1.3.4 Χώρα Β.1.4 Αριθμός τηλεφώνου: Β.1.5 Αριθμός fax: Β.1.6 e-mail: Β.2 ΝΟΜΙΜΟΣ ΕΚΠΡΟΣΩΠΟΣ9 ΤΟΥ ΧΟΡΗΓΟΥ ΣΤΗΝ ΚΟΙΝΟΤΗΤΑ ΓΙΑ ΤΟ ΣΚΟΠΟ ΤΗΣ ΠΑΡΟΥΣΑΣ ΔΟΚΙΜΗΣ (εάν είναι διαφορετικός από το χορηγό) Β.2.1 Επωνυμία οργανισμού: Β.2.2 Ονοματεπώνυμο υπεύθυνου επικοινωνίας: Β2.2.1 Όνομα Β2.2.2 Μεσαίο Όνομα Β2.2.3 Επίθετο Β.2.3 Διεύθυνση: Β2.3.1 Οδός Β2.3.2 Πόλη Β2.3.3 Ταχυδρομικός Κώδικας Β2.3.4 Χώρα Β.2.4 Αριθμός τηλεφώνου: Β.2.5 Αριθμός fax: Β.2.6 e-mail: Β.3 Β.3.1 Β.3.2 ΚΑΘΕΣΤΩΣ ΧΟΡΗΓΟΥ: Εμπορικός Μη εμπορικός Β.4 Πηγές χρηματικής ή υλικής υποστήριξης για την κλινική δοκιμή (επαναλάβετε αν είναι αναγκαίο): Επωνυμία οργανισμού: Χώρα: Β.4.1 Β.4.2 Β.5 Β.5.1 Β.5.2 Β5.3 Β5.3.1 Β5.3.2 Β5.3.3 Β5.3.4 Β.5.4 Β5.5 Β5.6 Σημείο επαφής10 ορισμένο από τον χορηγό για επιπλέον πληροφορίες για τη δοκιμή: Επωνυμία οργανισμού: Λειτουργικό όνομα σημείου επαφής (π.χ. Πίνακας Πληροφοριών Κλινικών Δοκιμών): Διεύθυνση: Οδός Πόλη Ταχυδρομικός Κώδικας Χώρα Αριθμός τηλεφώνου: Αριθμός fax: e-mail (χρησιμοποιήστε λειτουργική διεύθυνση για το ηλεκτρονικό ταχυδρομείο αντί για προσωπική) 9 Σύμφωνα με το άρθρο 19 της Οδηγίας 2001/20/EC Το σημείο επαφής θα πρέπει να δίνει λειτουργικές πληροφορίες παρά λεπτομέρειες για ένα άτομο, έτσι ώστε να αποφευχθεί η ανάγκη για επικαιροποίηση και διατήρηση αυτών των λεπτομερειών επαφής 10 Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 5 από 25 ΔΦΜΕ-E.4200-4/2 Γ. ΣΤΟΙΧΕΙΑ ΑΙΤΟΥΝΤΟΣ, (σημειώστε στο κατάλληλο τετραγωνίδιο) Γ.1 ΑΙΤΗΣΗ ΠΡΟΣ ΤΟΝ ΕΟΦ Γ.1.1 Χορηγός Γ.1.2 Νόμιμος εκπρόσωπος χορηγού Γ.1.3 Πρόσωπο ή οργανισμός εξουσιοδοτημένος από τον χορηγό να υποβάλει την αίτηση Γ.1.4 Συμπληρώστε παρακάτω τα στοιχεία του αιτούντος ακόμη και αν παρέχονται σε άλλο σημείο της αίτησης: Γ.1.4.1 Οργανισμός: Γ.1.4.2 Ονοματεπώνυμο υπεύθυνου επικοινωνίας: Γ.1.4.2.1 Όνομα Γ.1.4.2.2 Μεσαίο Όνομα Γ.1.4.2.3 Επίθετο Γ.1.4.3 Διεύθυνση: Γ.1.4.3.1 Οδός Γ.1.4.3.2 Πόλη Γ.1.4.3.3 Ταχυδρομικός Κώδικας Γ.1.4.3.4 Χώρα Γ.1.4.4 Αριθμός τηλεφώνου: Γ.1.4.5 Αριθμός fax: Γ.1.4.6 e-mail: Γ.1.5 Αίτηση για λήψη ενός .xml αντιγράφου των δεδομένων της αίτησης για κλινική δοκιμή (CTA data): Γ.1.5.1 Επιθυμείτε ένα .xml αντίγραφο των δεδομένων της CTA φόρμας, αποθηκευμένο στο EudraCT; Γ.1.5.1.1 Εάν ναι παραθέστε την(τις) ηλεκτρονική(-ές) διεύθυνση(-εις) (e-mail) στις οποίες θα πρέπει να αποσταλεί (έως 5 διευθύνσεις): Γ.1.5.1.2 Επιθυμείτε να το παραλάβετε μέσω e-mail συνδέσεων προστατευμένων με κωδικό;11 Εάν απαντήσετε όχι στην ερώτηση Γ.1.5.1.2 το xml αρχείο θα μεταδοθεί μέσω email συνδέσεων μειωμένης ασφάλειας ηλεκτρονικού ταχυδρομείου Γ.2 Γ.2.1 Γ.2.2 Γ.2.3 Γ.2.4 ΑΙΤΗΣΗ ΠΡΟΣ ΤΗΝ ΕΕΔ Χορηγός Νόμιμος εκπρόσωπος χορηγού Πρόσωπο ή οργανισμός εξουσιοδοτημένος από τον χορηγό να υποβάλει την αίτηση Ερευνητής υπεύθυνος για την αίτηση εάν υπάρχει:12 Ερευνητής συντονισμού (για πολυκεντρική δοκιμή) Κύριος ερευνητής (για μονοκεντρική δοκιμή) Συμπληρώστε παρακάτω τα στοιχεία του αιτούντος ακόμη και αν παρέχονται σε άλλο σημείο της αίτησης: Οργανισμός: Ονοματεπώνυμο υπεύθυνου επικοινωνίας: Όνομα Μεσαίο Όνομα Επίθετο Διεύθυνση: Οδός Πόλη Ταχυδρομικός Κώδικας Χώρα Αριθμός τηλεφώνου: Αριθμός fax: e-mail: Γ.2.5 Γ.2.5.1 Γ.2.5.2 Γ.2.5.2.1 Γ.2.5.2.2 Γ.2.5.2.3 Γ.2.5.3 Γ.2.5.3.1 Γ.2.5.3.2 Γ.2.5.3.3 Γ.2.5.3.4 Γ.2.5.4 Γ.2.5.5 Γ.2.5.6 ναι όχι ναι όχι Δ. ΠΛΗΡΟΦΟΡΙΕΣ ΓΙΑ ΚΑΘΕ ΥΠΟ ΕΡΕΥΝΑ ΦΑΡΜΑΚΕΥΤΙΚΟ ΠΡΟΪΟΝ (ΥΕΦΠ) 11 12 Απαιτείται ένας λογαριασμός EudraLink (βλέπε www.eudract.emea.eu.int για λεπτομέρειες) Σύμφωνα με την εθνική νομοθεσία Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 6 από 25 ΔΦΜΕ-E.4200-4/2 Στο παρόν τμήμα πρέπει να παρέχονται πληροφορίες για κάθε «χύμα (bulk) προϊόν» πριν από τις ειδικές προς τη δοκιμή ενέργειες (τυφλοποίηση, συσκευασία και επισήμανση) για κάθε υπό έρευνα φαρμακευτικό προϊόν (ΥΕΦΠ) που χρησιμοποιείται, συμπεριλαμβανομένου κάθε φαρμάκου σύγκρισης και κάθε εικονικού φαρμάκου, εάν απαιτείται. Για εικονικό φάρμακο πηγαίνετε στο Δ8. Εάν η δοκιμή διεξάγεται με αρκετά προϊόντα, χρησιμοποιήστε επιπρόσθετες σελίδες και αποδώστε σε κάθε προϊόν έναν διαδοχικό αριθμό στο Δ.1.1. Στην περίπτωση προϊόντος συνδυασμού, θα πρέπει να παρέχονται πληροφορίες για κάθε δραστικό συστατικό. Δ.1 ΠΡΟΣΔΙΟΡΙΣΜΟΣ ΤΟΥ ΥΕΦΠ Υποδείξτε ποιο από τα παρακάτω περιγράφεται και ακολούθως επαναλάβετε, όπως απαιτείται, για έκαστο αριθμημένο ΥΕΦΠ που πρόκειται να χρησιμοποιηθεί στη δοκιμή (αριθμήστε από 1-n): Δ.1.1 Το παρόν αναφέρεται στον αριθμό του ΥΕΦΠ: (..) Δ.1.2 ΥΕΦΠ υπό δοκιμή Δ.1.3 ΥΕΦΠ χρησιμοποιούμενο ως φάρμακο σύγκρισης Δ.2 Δ.2.1 Δ.2.1.1 Δ.2.1.1.1 Δ.2.1.1.1.1 Δ.2.1.1.2 Δ.2.1.1.3 Δ.2.1.1.4 Δ.2.1.1.4.1 Δ.2.1.2 Δ.2.1.2.1 Δ.2.2 Δ.2.2.1 Δ.2.2.1.1 Δ.2.2.2 ΚΑΘΕΣΤΩΣ ΤΟΥ ΥΕΦΠ Υπάρχει άδεια κυκλοφορίας για το ΥΕΦΠ που πρόκειται να χρησιμοποιηθεί στη δοκιμή; ναι όχι Αν το ΥΕΦΠ έχει άδεια κυκλοφορίας στο Κράτος Μέλος που αφορά η αίτηση αλλά το εμπορικό όνομα και ο κάτοχος άδειας κυκλοφορίας δεν έχουν οριστεί στο πρωτόκολλο, πηγαίνετε στο τμήμα Δ..2.2 Εάν ναι στο Δ.2.1, προσδιορίστε το προϊόν που πρόκειται να χρησιμοποιηθεί στη δοκιμή: Εμπορική ονομασία:13 EV Κωδικός Προϊόντος(όπου χρειάζεται): Επωνυμία κατόχου ΑΚ: Αριθμός ΑΚ (εάν η ΑΚ εκδόθηκε από Κράτος Μέλος): Το ΥΕΦΠ έχει τροποποιηθεί σε σχέση με την ΑΚ του; ναι όχι Εάν ναι, προσδιορίστε: Ποια χώρα χορήγησε την ΑΚ; (……..) Είναι αυτό το Κράτος Μέλος στο οποίο αφορά η παρούσα αίτηση; ναι όχι Περιπτώσεις όπου υπάρχει ΑΚ στο οικείο Κράτος Μέλος για το ΥΕΦΠ που πρόκειται να χρησιμοποιηθεί στην κλινική δοκιμή, αλλά το πρωτόκολλο επιτρέπει τη χορήγηση στους συμμετέχοντες στη δοκιμή οιουδήποτε σήματος του ΥΕΦΠ για το οποίο υπάρχει ΑΚ στο οικείο κράτος μέλος και ο προσδιορισμός του/των ΥΕΦΠ(ων) πριν από την έναρξη της δοκιμής δεν είναι εφικτός Στο πρωτόκολλο, η θεραπεία ορίζεται μόνο με βάση το δραστικό συστατικό; ναι όχι Εάν ναι, δηλώστε το δραστικό συστατικό στο Δ.3.8 ή Δ.3.9 ναι όχι Στο πρωτόκολλο, η θεραπεία επιτρέπει διάφορους συνδυασμούς προϊόντων που κυκλοφορούν στο εμπόριο χρησιμοποιούμενους σύμφωνα με την τοπική κλινική πρακτική σε ορισμένα ή όλα τα ερευνητικά κέντρα του Κράτους Μέλους; Δ.2.2.2.1 Δ.2.2.3 Δ.2.2.4 Δ.2.2.4.1 Δ.2.3 Δ.2.3.1 Εάν ναι , δηλώστε το δραστικό συστατικό στο Δ.3.8 ή Δ.3.9 Τα προϊόντα που πρόκειται να χορηγηθούν ως ΥΕΦΠ ορίζεται ότι ανήκουν σε ομάδα ATC;6 Εάν ναι, αναφέρατε την ομάδα ATC των εφαρμοστέων εγκεκριμένων κωδικών στο πεδίο κωδικών ATC (επίπεδο 3 ή ανώτερο στο βαθμό που μπορεί να προσδιοριστεί) στο Δ.3.3 Άλλο: Εάν ναι, παρακαλώ προσδιορίστε: Φάκελος Υπό Έρευνα Φαρμακευτικού Προϊόντος (IMPD) που υποβλήθηκε: Πλήρης IMPD Δ.2.3.2 Δ.2.3.3 Δ.2.4 Δ.2.2.3.1 13 ναι όχι ναι όχι ναι όχι Απλοποιημένος IMPD Περίληψη των Χαρακτηριστικών του Προϊόντος (SmPC) μόνο ναι ναι όχι όχι Υπάρχει παλαιότερη έγκριση για το ΥΕΦΠ στο πλαίσιο κλινικής δοκιμής ναι όχι Ο κωδικός ATC αναφέρεται στην Περίληψη Χαρακτηριστικών του Προϊόντος (SmPC) Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 7 από 25 ΔΦΜΕ-E.4200-4/2 Δ.2.4.1 Δ.2.5 Δ.2.5.1 Δ.2.6 Δ.2.6.1 Δ.2.6.1.1 Δ.2.6.1.2 που διενήργησε ο χορηγός στην Κοινότητα; Εάν ναι, προσδιορίστε σε ποια Κράτη Μέλη: Έχει χαρακτηριστεί το ΥΕΦΠ στην παρούσα ένδειξη ως ορφανό φάρμακο στην Κοινότητα ; Εάν ναι, αναφέρατε τον αριθμό χαρακτηρισμού του φαρμάκου ως ορφανού14 Έχει αποτελέσει το ΥΕΦΠ αντικείμενο επιστημονικής συμβουλής (scientific advise) που να σχετίζεται με την παρούσα κλινική δοκιμή; Εάν ναι στο Δ.2.6 υποδείξτε την πηγή της συμβουλής και παραθέστε ένα αντίγραφο στην αίτηση της κλινικής δοκιμής: Από την CHMP;15 Από την αρμόδια αρχή ενός Κράτους Μέλους; Δ.3 Δ.3.1 Δ.3.2 Δ.3.3 ΠΕΡΙΓΡΑΦΗ ΤΟΥ ΥΕΦΠ Ονομασία προϊόντος εάν υπάρχει16: Κωδικός προϊόντος εάν υπάρχει17: Κωδικός ATC, εάν υπάρχει επίσημη καταχώρηση18: Δ.3.4 Δ.3.4.1 Φαρμακοτεχνική μορφή (χρησιμοποιήστε τυποποιημένους όρους): Πρόκειται για ειδική παιδιατρική σύνθεση? ναι όχι Μέγιστη διάρκεια θεραπείας ενός συμμετέχοντα σύμφωνα με το πρωτόκολλο: Δ.3.5 Δ.3.6 Δ.3.6.1 όχι ναι ( ) όχι ναι ναι όχι όχι Μέγιστη επιτρεπόμενη δόση : Πρώτη δόση για κλινική δοκιμή όπου χορηγείται πρώτη φορά σε ανθρώπους(προσδιορίστε: ανά ημέρα ή ολική δόση, μονάδες και οδός χορήγησης) Μέγιστη επιτρεπόμενη δόση (προσδιορίστε: ανά ημέρα ή ολική δόση, μονάδες και οδός χορήγησης) Δ.3.6.2 1.1.1 ναι Δ.3.7 Οδός χορήγησης (χρησιμοποιήστε τυποποιημένους όρους): Δ.3.8 Ονομασία κάθε δραστικού συστατικού [INN (Κοινόχρηστη Διεθνής Ονομασία) ή προτεινόμενη INN εάν υπάρχει]: Δ.3.9 Δ.3.9.1 Δ.3.9.2 Δ.3.9.3 Δ.3.9.4 Δ.3.9.5 Δ.3.9.6 Άλλη διαθέσιμη ονομασία για κάθε δραστικό συστατικό (αναφέρετε όλα τα διαθέσιμα στοιχεία): Αριθμός CAS19 Τρέχων Κωδικός Χορηγού Άλλο περιγραφικό όνομα EV Κωδικός Ουσίας Πλήρης Μοριακός Τύπος Χημική/Βιολογική περιγραφή της δραστικής ουσίας Δ.3.10 Δ.3.10.1 Δ.3.10.2 Δ.3.10.3 Περιεκτικότητα (προσδιορίστε όλες τις περιεκτικότητες που πρόκειται να χρησιμοποιηθούν): Μονάδα συγκέντρωσης: Τύπος συγκέντρωσης (“ακριβής αριθμός”, “εύρος”, “περισσότερο από” ή “έως και”) Συγκέντρωση (αριθμός): 14 0 Σύμφωνα με την καταχώρηση της Κοινότητας για τα ορφανά φάρμακα (Κανονισμός (EC) n 141/2000): http://ec.europa.eu/enterprise/pharmaceuticals/register/index.htm 15 Committee for Medicinal Products for Human Use of the European Medicines Agency 16 Να αναφέρεται μόνο όταν δεν υπάρχει εμπορικό όνομα. Πρόκειται για το όνομα που χρησιμοποιείται συνηθέστατα από το χορηγό για τον προσδιορισμό του ΥΕΦΠ στην τεκμηρίωση της κλινικής δοκιμής (πρωτόκολλο, ενημερωτικό φυλλάδιο για τον ερευνητή…) 17 Να αναφέρεται μόνο όταν δεν υπάρχει εμπορικό όνομα. Πρόκειται για τον κωδικό που καθορίζεται από το χορηγό και αντιπροσωπεύει το όνομα που χρησιμοποιείται συνηθέστατα για τον προσδιορισμό του ΥΕΦΠ στην τεκμηρίωση της κλινικής δοκιμής. Για παράδειγμα ένας κωδικός μπορεί να χρησιμοποιείται για συνδυασμούς φαρμάκων ή φαρμάκων και ιατροτεχνολογικών προϊόντων. 18 Αναφέρεται στην Περίληψη Χαρακτηριστικών του Προϊόντος (SmPC) 19 Chemical Abstracts Service Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 8 από 25 ΔΦΜΕ-E.4200-4/2 Δ.3.11 Τύπος ΥΕΦΠ Το ΥΕΦΠ περιέχει ένα δραστικό συστατικό: Δ.3.11.1 χημικής προέλευσης; Δ.3.11.2 βιολογικής / βιοτεχνολογικής προέλευσης(διαφορετικό από ΥΕΦΠ Προηγμένων Θεραπειών); Πρόκειται για: Δ.3.11.3 ΥΕΦΠ Προηγμένης Θεραπείας (ATIMP); Δ.3.11.3.1 Φάρμακο σωματοκυτταρικής θεραπείας; 20 Δ.3.11.3.2 Φάρμακο γονιδιακής θεραπείας;21 Δ.3.11.3.3 Προϊόν Μηχανικής Ιστού(Τissue engineered product)22 Δ.3.11.3.4 Συνδυασμός ΥΕΦΠ Προηγμένης Θεραπείας(π.χ. που συμπεριλαμβάνει ιατροτεχνολογικό προϊόν)23 Δ3.11.3.5 Έχει εκδώσει η Επιτροπή για Προηγμένες Θεραπείες ταξινόμηση για αυτό το προϊόν ; Δ3.11.3.5. Αν ναι, αναφέρατε αυτή την ταξινόμηση και τον αριθμό αναφοράς: 1 Προϊόν συνδυασμού που περιλαμβάνει ιατροτεχνολογικό προϊόν αλλά όχι Δ3.11.4 Προηγμένης Θεραπείας ; Δ.3.11.5 Ραδιοφαρμακευτικό προϊόν; Δ.3.11.6 Ανοσολογικό φαρμακευτικό προϊόν (όπως εμβόλιο, αλλεργιογόνο, ανοσοποιητικός ορός); Δ.3.11.7 Φαρμακευτικό προϊόν παραγώγου πλάσματος; Δ.3.11.8 Φαρμακευτικό προϊόν εκχύλισης; Δ.3.11.9 Ανασυνδιασμένο φαρμακευτικό προϊόν; Δ.3.11.10 Φαρμακευτικό προϊόν που περιέχει γενετικά τροποποιημένους οργανισμούς; Δ.3.11.10. Έχει χορηγηθεί άδεια για περιορισμένη χρήση ή αποδέσμευση; 1 Δ.3.11.10.2 Μήπως εκκρεμεί; Δ.3.11.11. Φυτικό φαρμακευτικό προϊόν; Δ.3.11.12 Ομοιοπαθητικό φαρμακευτικό προϊόν; Δ.3.11.13 Άλλος τύπος φαρμακευτικού προϊόντος; Δ.3.11.13. Εάν ναι, προσδιορίστε:; 1 Τρόπος δράσης (Ελεύθερο κείμενο) 24 Δ 3.12 Πρόκειται για ΥΕΦΠ που θα χρησιμοποιηθεί σε κλινική δοκιμή για πρώτη φορά Δ 3.13 σε ανθρώπους ; Δ 3.13.1 Δ.4 Δ.4.1 Δ.4.1.1 Δ.4.1.2 Δ.4.1.3 Δ.4.1.3.1 Δ.4.2 Δ.4.2.1 Δ.4.2.2 Δ.4.2.2.1 Δ.4.2.3 Δ.4.2.3.1 ναι ναι ναι ναι ναι ναι όχι όχι όχι ναι όχι όχι όχι όχι ναι όχι ναι ναι ναι όχι όχι όχι ναι ναι ναι ναι ναι όχι όχι όχι όχι όχι ναι ναι ναι όχι όχι όχι Εάν ναι, έχουν ταυτοποιηθεί οι παράγοντες κινδύνου σύμφωνα με την Οδηγία (FIH)25 ; ΥΠΟ ΕΡΕΥΝΑ ΦΑΡΜΑΚΕΥΤΙΚΟ ΠΡΟΪΟΝ ΣΩΜΑΤΟΚΥΤΤΑΡΙΚΗΣ ΘΕΡΑΠΕΙΑΣ (ΠΟΥ ΔΕΝ ΕΧΕΙ ΥΠΟΣΤΕΙ ΓΕΝΕΤΙΚΗ ΤΡΟΠΟΠΟΙΗΣΗ) Προέλευση των κυττάρων Αυτόλογη ναι όχι Αλλογονιδιακή ναι όχι Ξενογονιδιακή ναι όχι Εάν ναι, προσδιορίστε το είδος προέλευσης: Τύπος κυττάρων Βλαστικά κύτταρα ναι όχι Διαφοροποιημένα κύτταρα ναι όχι Εάν ναι, προσδιορίστε τον τύπο (π.χ. κερατινοκύτταρα, ινοβλάστες, χονδροκύτταρα,…): Άλλο: ναι όχι Εάν άλλο, προσδιορίστε: 20 Συμπληρώστε επίσης το Δ4 για την σωματοκυτταρική θεραπεία όπως ορίζεται στο Annex 1 part IVτης Directive 2001/83/EC όπως έχει τροποποιηθεί. 21 Συμπληρώστε επίσης το Δ5 για την γονιδιακή θεραπεία όπως ορίζεται στο Annex 1 part IVτης Directive 2001/83/EC όπως έχει τροποποιηθεί. 22 Συμπληρώστε επίσης το Δ6 – Προϊόντα Μηχανικής Ιστών όπως ορίζονται στο άρθρο 2(1)(b) του Κανονισμού 1394/2007/EC. 23 Συμπληρώστε επίσης το Δ.7 24 Ο τρόπος δράσης θα πρέπει εν συντομία να περιγράφει τις χημικές, βιοχημικές, ανοσολογικές ή βιολογικές ιδιότητες που εμφανίζει το ΥΕΦΠ για να πραγματοποιήσει την φαρμακολογική του δράση. 25 Guideline on strategies to identify and mitigate risks for first-in-human clinical trials with investigational medicinal products. EMEA/CHMP/SWP/28367/2007 19 July 2007 Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 9 από 25 ΔΦΜΕ-E.4200-4/2 Δ.5 Δ.5.1 Δ.5.2 Δ.5.3 Δ.5.4 Δ.5.4.1 Δ.5.4.1.1 Δ.5.4.1.2 Δ.5.4.2 Δ.5.4.2.1 Δ.5.4.3 Δ.5.4.3.1 Δ.5.5 ΥΠΟ ΕΡΕΥΝΑ ΦΑΡΜΑΚΕΥΤΙΚΑ ΠΡΟΪΟΝΤΑ ΓΟΝΙΔΙΑΚΗΣ ΘΕΡΑΠΕΙΑΣ Εξεταζόμενο(α) γονίδιο(α): Γονιδιακή θεραπεία in vivo: Γονιδιακή θεραπεία ex vivo: Τύπος προϊόντος από μεταφορά γονιδίων Νουκλεϊνικό οξύ (λ.χ. πλασμίδιο): Εάν ναι, προσδιορίστε: Γυμνό Συμπλοκοποιημένο Ιογενής φορέας Εάν ναι, προσδιορίστε τον τύπο: αδενοϊός, ρετροϊός, AAV, …: Άλλα: Εάν άλλα, προσδιορίστε: ναι ναι όχι όχι ναι όχι ναι ναι ναι όχι όχι όχι ναι όχι ναι όχι ναι ναι ναι όχι όχι όχι ναι όχι Δ.5.5.1 Δ.5.5.2 Δ.5.5.3 Δ.5.5.3.1 Δ.5.5.4 Γενετικά τροποποιημένα κύτταρα: Εάν ναι, προσδιορίστε την προέλευση των κυττάρων: Αυτόλογη: Αλλογονιδιακή: Ξενογονιδιακή: Εάν ναι, προσδιορίστε το είδος προέλευσης: Προσδιορίστε τον τύπο κυττάρων (βλαστικά αιμοποιητικά κύτταρα, …): Δ.6 ΠΡΟΙΟΝ ΜΗΧΑΝΙΚΗΣ ΙΣΤΟΥ Δ.6.1 Δ.6.1.1 Δ.6.1.2 Δ.6.1.3 Δ.6.1.3.1 Δ.6.2 Δ.6.2.1 Δ.6.2.2 Δ.6.2.2.1 Δ.6.2.3 Δ.6.2.3.1 Η ένδειξη η οποία καθορίζει ότι πρόκειται για προϊόν μηχανικής ιστού εν αντιθέσει με προϊόν προερχόμενο από κυτταρική θεραπεία δίνεται στην ενότητα Ε1.1. Προέλευση των κυττάρων Αυτόλογη ναι όχι Αλλογονιδιακή ναι όχι Ξενογονιδιακή ναι όχι Εάν ναι, προσδιορίστε το είδος προέλευσης: Τύπος κυττάρων Βλαστικά κύτταρα ναι όχι Διαφοροποιημένα κύτταρα ναι όχι Εάν ναι, προσδιορίστε τον τύπο (π.χ. κερατινοκύτταρα, ινοβλάστες, χονδροκύτταρα,…): Άλλο: ναι όχι Εάν άλλο, προσδιορίστε: Δ.7 Δ.7.1 Δ.7.2 Δ.7.3 Δ.7.4 Δ.7.4.1 Δ.7.4.1.1 Δ.7.1.1.1 Δ.7.4.2 Δ.7.4.3 Δ.7.4.4 Δ.7.4.5 Δ 7.4.5.1 ΠΡΟΙΟΝΤΑ ΠΟΥ ΠΕΡΙΕΧΟΥΝ ΣΥΣΚΕΥΕΣ(Π.Χ. ΙΑΤΡΟΤΕΧΝΟΛΟΓΙΚΑ, ΙΚΡΙΩΜΑΤΑ ΚΛΠ ) Σύντομη περιγραφή της συσκευής: Ονομασία συσκευής: Είναι εμφυτεύσιμη συσκευή ; ναι όχι Το προϊόν περιέχει: Ιατροτεχνολογικό προϊόν; ναι όχι Το Ιατροτεχνολογικό προϊόν διαθέτει CE σήμανση ; ναι όχι O οργανισμός διαπίστευσης είναι: Βιο-υλικά ; ναι όχι Ικριώματα ; ναι όχι Μήτρες ; ναι όχι Άλλο ; ναι όχι Αν πρόκειται για άλλο, προσδιορίστε: Δ.8 ΠΛΗΡΟΦΟΡΙΕΣ ΣΧΕΤΙΚΑ ΜΕ ΤΟ ΕΙΚΟΝΙΚΟ ΦΑΡΜΑΚΟ (εφόσον συντρέχει περίπτωση; επαναλάβετε αν είναι απαραίτητο) Χρησιμοποιείται εικονικό φάρμακο; ναι όχι Αναφέρεται στον αριθμό του εικονικού φαρμάκου: (..) Δ.8.1 Δ.8.2 Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 10 από 25 ΔΦΜΕ-E.4200-4/2 Δ.8.3 Δ.8.4 Δ.8.5 Δ.8.5.1 Δ.8.5.2 Δ.8.5.2.1 Φαρμακοτεχνική μορφή: Οδός χορήγησης: Ποιο ΥΕΦΠ αντιστοιχεί στο εικονικό φάρμακο; Αναφέρατε Αριθμό(-ούς) ΥΕΦΠ από το Δ.1.1 (..) Σύνθεση, εκτός του(των) δραστικού(-ών) συστατικού(-ών): Είναι κατά τα άλλα όμοιο με το ΥΕΦΠ; Εάν όχι, προσδιορίστε τα βασικά συστατικά: ναι όχι Δ.9 ΚΕΝΤΡΟ(Α) ΟΠΟΥ ΤΟ ΕΙΔΙΚΕΥΜΕΝΟ ΠΡΟΣΩΠΟ ΠΙΣΤΟΠΟΙΕΙ ΤΗΝ ΑΠΟΔΕΣΜΕΥΣΗ ΤΗΣ ΠΑΡΤΙΔΑ26 Το παρόν τμήμα αφορά τα τελικά ΥΕΦΠ, ήτοι τα φαρμακευτικά προϊόντα που έχουν τυχαιοποιηθεί, συσκευαστεί, επισημανθεί και πιστοποιηθεί για να χρησιμοποιηθούν στην κλινική δοκιμή. Όταν πρόκειται για πολλά κέντρα ή πιστοποιηθούν περισσότερα από ένα ΥΕΦΠ χρησιμοποιείστε επί πλέον σελίδες και δώστε σε κάθε ΥΕΦΠ τον αριθμό του από το τμήμα Δ.1.1 ή Δ.8.2. Σε περίπτωση πολλών κέντρων υποδείξετε το προϊόν που πιστοποιείται από κάθε κέντρο. Δ.9.1 Μην συμπληρώσετε το τμήμα Δ.9.2 για ένα ΥΕΦΠ το οποίο: - Διαθέτει Άδεια Κυκλοφορίας στην Ε.Ε και - Προέρχεται από την την Ε.Ε και - Χρησιμοποιείται στη δοκιμή χωρίς μετατροπή (π.χ. όχι υπερενκαψυλίωση) και - Η συσκευασία και η επισήμανση γίνεται μόνο για τοπική χρήση σύμφωνα με το Άρθρο 9.2 της Οδηγίας 2005/28/EC (Οδηγία GCP) Εφόσον πληρούνται όλοι αυτοί οι όροι μαρκάρετε το τετραγωνίδιο και αναφέρετε τον(τους) αριθμό(ούς) για κάθε ΥΕΦΠ συμπεριλαμβανομένου του εικονικού φαρμάκου από τα τμήματα Δ.1.1 και Δ.8.2 στα οποία αναφέρονται: (..) Ποιος είναι αρμόδιος στην Κοινότητα για την πιστοποίηση του τελικού ΥΕΦΠ; Το τμήμα αυτό είναι αρμόδιο για την πιστοποίηση των (αναφέρετε τον/τους αριθμό/ούς για κάθε ΥΕΦΠ συμπεριλαμβανομένου του εικονικού φαρμάκου από τα τμήματα Δ.1.1 και Δ.8.2): (..); Δ.9.2 σημειώστε στο κατάλληλο τετραγωνίδιο : Δ.9.2.1 Δ.9.2.2 Παρασκευαστής Εισαγωγέας Δ.9.2.3 Δ.9.2.4 Δ.9.2.4.1 Δ.9.2.4.2 Δ.9.2.4.3 Δ.9.2.4.4 Επωνυμία οργανισμού: Διεύθυνση: Δ.9.2.5 Δ.9.2.5.1 Αναφέρατε τον αριθμό εγκρίσεως του παρασκευαστή: Εάν δεν υπάρχει έγκριση, εξηγήστε τους λόγους: Οδός Πόλη Ταχυδρομικός Κώδικας Χώρα Σε περίπτωση που το προϊόν δεν έχει άδεια κυκλοφορίας στην Ε.Ε, αλλά παρέχεται χύμα και η τελική συσκευασία και επισήμανση για τοπική χρήση γίνεται σύμφωνα με το Άρθρο 9.2 της Οδηγίας 2005/28/EC (Οδηγία GCP), τότε συμπληρώστε στο Δ.9.2 το κέντρο όπου το προϊόν είχε τελικά πιστοποιηθεί για αποδέσμευση από το ειδικευμένο πρόσωπο προς χρήση στην κλινική δοκιμή Ε. ΓΕΝΙΚΕΣ ΠΛΗΡΟΦΟΡΙΕΣ ΓΙΑ ΤΗ ΔΟΚΙΜΗ Το τμήμα αυτό θα πρέπει να χρησιμοποιείται για την παροχή πληροφοριών σχετικά με τους στόχους, τον σκοπό και το σχεδιασμό της δοκιμής. Όταν το πρωτόκολλο περιλαμβάνει μια υπο-δοκιμή που αφορά στο Κράτος Μέλος,, 26 Σύμφωνα με την παράγραφο 38 του Παραρτήματος 13 του ‘Volume 4 of the Rules Governing Medicinal Products in the European Union’ Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 11 από 25 ΔΦΜΕ-E.4200-4/2 θα πρέπει να συμπληρώνεται το τμήμα Ε.2.3 παρέχοντας πληροφορίες για την υπο-δοκιμή. Για να την προσδιορίσετε σημειώστε στο τετραγωνίδιο για την υπο-δοκιμή στην ερώτηση «Στόχος της δοκιμής» παρακάτω. Ε.1 Ε.1.1 Ε.1.1.1 Ε.1.1.2 Ε.1.2 Ε.1.3 ΙΑΤΡΙΚΗ ΚΑΤΑΣΤΑΣΗ Ή ΑΣΘΕΝΕΙΑ ΥΠΟ ΕΡΕΥΝΑ Προσδιορίστε την ιατρική κατάσταση/σεις που θα ερευνηθεί (ελεύθερο κείμενο): 27 Ιατρική κατάσταση σε απλή κατανοητή γλώσσα Θεραπευτικό Πεδίο Έκδοση, επίπεδο, όρος και κωδικός ταξινόμησης του MedDRA (επαναλάβετε όπου είναι απαραίτητο):28 Πρόκειται κάποια από τις ιατρικές καταστάσεις να δοκιμασθεί ως σπάνια ασθένεια;29 ναι όχι ναι όχι Ε.2 Ε.2.1 Ε.2.2 Ε.2.3 Ε.2.3.1 ΣΤΟΧΟΣ ΤΗΣ ΔΟΚΙΜΗΣ Κύριος στόχος: Δευτερεύοντες στόχοι: Υπάρχει κάποια υπο-δοκιμή; Εάν ναι δώστε τον πλήρη τίτλο, ημερομηνία και έκδοση κάθε υπο-δοκιμής και τους σχετιζόμενους με αυτήν στόχους: Ε.3 ΚΥΡΙΑ ΚΡΙΤΗΡΙΑ ΕΝΤΑΞΗΣ (αναφέρετε τα σημαντικότερα) Ε.4 ΚΥΡΙΑ ΚΡΙΤΗΡΙΑ ΑΠΟΚΛΕΙΣΜΟΥ (αναφέρετε τα σημαντικότερα) Ε.5 Ε.5.1 Ε.5.1.1 Ε.5.2 Ε.5.2.1 Τελικό(-ά) σημείο(-α): Πρωτεύον Τελικό Σημείο(επαναλάβετε αν χρειάζεται)30 Χρονικό σημείο εκτίμησης αυτού του τελικού σημείου Δευτερεύον τελικό σημείο (επαναλάβετε αν χρειάζεται) Χρονικό σημείο εκτίμησης αυτού του τελικού σημείου Ε.6 Ε.6.1 Ε.6.2 Ε.6.3 Ε.6.4 Ε.6.5 Ε.6.6 Ε.6.7 Ε.6.8 Ε.6.9 Ε.6.10 Ε.6.11 Ε.6.12 Ε.6.13 Ε.6.13.1 ΣΚΟΠΟΣ ΤΗΣ ΔΟΚΙΜΗΣ – Σημειώστε σε όλα τα κατάλληλα τετραγωνίδια, όπου συντρέχει περίπτωση Διάγνωση Προφύλαξη Θεραπεία Ασφάλεια Αποτελεσματικότητα Φαρμακοκινητική Φαρμακοδυναμική Βιοϊσοδυναμία Δόση-Απάντηση Φαρμακογενετική Φαρμακογονιδιωματική Φαρμακοοικονομική Άλλο Εάν άλλο, προσδιορίστε: Ε.7 Ε.7.1 ΤΥΠΟΣ ΔΟΚΙΜΗΣ31 Φαρμακολογική δοκιμή σε ανθρώπους (Φάση I) Πρόκειται για: Πρώτη χορήγηση σε ανθρώπους Ε.7.1.1 27 Σε περιπτώσεις δοκιμών σε υγιείς εθελοντές θα πρέπει να αναφέρεται η ένδειξη που προορίζεται για το υπό ανάπτυξη προϊόν. 28 Οι αιτούντες θα πρέπει να ενθαρρύνονται στην χρήση του ‘MedDRA lower level term if applicable and classification code’. Σχετική πρόσβαση στοEMEA EudraCT website (http://eudract.emea.europa.eu/). 29 Σημεία που πρέπει να ληφθούν υπόψη κατά τον υπολογισμό και την αναφορά του επιπολασμού μιας ιατρικής κατάστασης για τον χαρακτηρισμό ενός Ορφανού φαρμάκου: COM/436/01 (http://www.emea.europa.eu/htms/human/orphans/intro.htm). 30 Το πρωτόκολλο συνήθως προσδιορίζει ένα Πρωτεύον Τελικό Σημείο , όμως μπορεί να υπάρχουν περισσότερα και σε μερικές περιπτώσεις και/η αριθμός Δευτερευόντων Τελικών Σημείων. 31 Οι περιγραφές των τύπων των δοκιμών που παρέχονται , είναι κατά προτίμηση αυτές που συνίστανται σε Φάσεις. Βλέπε σελ. 5 of Community guideline CPMP/ICH/291/95. Η ανάπτυξη μίας νέας ένδειξης μετά την αρχική έγκριση ενός φαρμάκου θεωρείται νέο σχέδιο ανάπτυξης. Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 12 από 25 ΔΦΜΕ-E.4200-4/2 Ε.7.1.2 Ε.7.1.3 Ε.7.1.3.1 Ε.7.2 Ε.7.3 Ε.7.4 Μελέτη βιοϊσοδυναμίας Άλλο: Εάν άλλο, προσδιορίστε: Θεραπευτική διερευνητική (Φάση II) Θεραπευτική επιβεβαίωσης (Φάση III) Θεραπευτική χρήση (Φάση IV) Ε.8 Ε.8.1 ΣΧΕΔΙΑΣΜΟΣ ΤΗΣ ΔΟΚΙΜΗΣ Ελεγχόμενη Εάν ναι, προσδιορίστε: Ε.8.1.1 Τυχαιοποιημένη Ε.8.1.2 Ανοιχτή: Ε.8.1.3 Μονή τυφλή: Ε.8.1.4 Διπλή τυφλή: Ε.8.1.5 Παράλληλη ομάδα: Ε.8.1.6 Διασταυρούμενη: Ε.8.1.7 Άλλο: Ε.8.1.7.1 Εάν ναι στο 8.1.7, προσδιορίστε: Ε.8.2 Εάν ελεγχόμενη, προσδιορίστε το φάρμακο σύγκρισης: Ε.8.2.1 Άλλο(α) φάρμακο(α) Ε.8.2.2 Εικονικό φάρμακο Ε.8.2.3 Άλλο Ε.8.2.3.1 Εάν ναι, στο άλλο, προσδιορίστε: Ε.8.2.4 Αριθμός σκελών στην δοκιμή Ε.8.3 Ένα κέντρο στο Ενεχόμενο Κράτος Μέλος (βλέπε επίσης τμήμα Ζ): Ε.8.4 Πολλά κέντρα στο Ενεχόμενο Κράτος Μέλος (βλέπε επίσης τμήμα Ζ): Ε.8.4.1 Αναμενόμενος αριθμός κέντρων στο Ενεχόμενο Κράτος Μέλος ( ) Ε.8.5 Πολλά Κράτη Μέλη: Αναμενόμενος αριθμός κέντρων στην Κοινότητα ( ) 1.1.2 Ε.8.5.1 Ε.8.6 Ε.8.6.1 Ε.8.6.2 Ε.8.6.3 Ε.8.6.4 Ε.8.7 Ε.8.8 Ε.8.9 Ε.8.9.1 Ε.8.9.2 Ε.8.10 Ε.8.10.1 Ε.8.10.2 Η δοκιμή αφορά κέντρα εκτός ΕΟΧ Η δοκιμή διεξάγεται σε κέντρα εντός και εκτός ΕΟΧ Η δοκιμή διεξάγεται σε κέντρα εντελώς εκτός ΕΟΧ Αν το Ε.8.6.1 ή το Ε.8.6.2 είναι ναι, προσδιορίστε τις περιοχές όπου σχεδιάζονται κέντρα(επαναλάβετε αν χρειάζεται:) Αν το Ε.8.6.1 ή το Ε.8.6.2 είναι ναι, προσδιορίστε τον αριθμό των κέντρων που προβλέπονται εκτός ΕΟΧ ναι όχι ναι ναι ναι ναι ναι ναι ναι όχι όχι όχι όχι όχι όχι όχι ναι ναι ναι όχι όχι όχι ναι ναι όχι όχι ναι όχι ναι όχι ναι ναι όχι όχι ναι όχι Υπάρχει ανεξάρτητη επιτροπή παρακολούθησης των δεδομένων της δοκιμής; Προσδιορισμός της λήξης της δοκιμής: Εάν ορίζεται ως η τελευταία επίσκεψη του τελευταίου συμμετέχοντος σημειώσατε “LVLS”. Εάν όχι δώστε τον ορισμό: Αρχική εκτίμηση της διάρκειας της δοκιμής32 (έτη, μήνες και ημέρες) : Στο ενεχόμενο Κράτος Μέλος έτη μήνες ημέρες Σε όλες τις ενεχόμενες χώρες από τη δοκιμή έτη μήνες ημέρες Προτεινόμενη ημερομηνία έναρξης της στρατολόγησης Στο ενεχόμενο Κράτος Μέλος Σε οποιοδήποτε Κράτος Μέλος ΣΤ. ΠΛΗΘΥΣΜΟΣ ΤΩΝ ΣΥΜΜΕΤΕΧΟΝΤΩΝ ΣΤΗ ΔΟΚΙΜΗ ΣΤ.1 ΗΛΙΚΙΑΚΟ ΕΥΡΟΣ ΣΤ.1.1 Κάτω των 18 ετών Εάν ναι, προσδιορίστε τον εκτιμώμενο αριθμό συμμετεχόντων που σχεδιάζεται για κάθε ηλικία για όλη τη δοκιμή: Aρ. συμμετεχόντων κατά προσέγγιση33: ναι όχι 32 Από την πρώτη ένταξη έως την τελευταία επίσκεψη του τελευταίου συμμετέχοντος. Αυτοί οι αριθμοί αποτελούν αρχικές εκτιμήσεις. Δεν είναι απαραίτητο οι αιτούντες να επικαιροποιούν αυτή την πληροφορία ούτε συνιστάται έγκριση ή περιορισμός στην εισαγωγή αυτών των ασθενών στην δοκιμή. Ο αριθμός των συμμετεχόντων των 33 Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 13 από 25 ΔΦΜΕ-E.4200-4/2 Εάν ναι, προσδιορίστε: ΣΤ.1.1.1 In Utero (στη Μήτρα) ΣΤ.1.1.2 Πρόωρα νεογνά (ηλικία κύησης < 37 εβδομάδες) ΣΤ.1.1.3 Νεογέννητα (0-27 ημερών) ΣΤ.1.1.4 Βρέφη και νήπια (28 ημερών - 23 μηνών) ΣΤ.1.1.5 Παιδιά (2-11 ετών) ΣΤ.1.1.6 Έφηβοι (12-17 ετών) ΣΤ.1.2 Ενήλικες (18-64 ετών) ΣΤ.1.3 Ηλικιωμένοι (> 65= ετών) ΣΤ.2 ΣΤ.2.1 ΣΤ.2.2 ΣΤ.3 ΣΤ.3.1 ΣΤ.3.2 ΣΤ.3.3 ΣΤ.3.3.1 ναι ναι ναι ναι ναι ναι ναι ναι όχι όχι όχι όχι όχι όχι όχι όχι ναι ναι ναι ναι όχι όχι όχι όχι ναι ναι ναι ναι ναι όχι όχι όχι όχι όχι ναι όχι ΦΥΛΟ Θηλυκό Αρσενικό ΣΤ.3.3.2 ΣΤ.3.3.3 ΣΤ.3.3.4 ΣΤ.3.3.5 ΣΤ.3.3.6 ΣΤ.3.3.6.1 ΣΤ.3.3.7 ΣΤ.3.3.7.1 ΟΜΑΔΕΣ ΣΥΜΜΕΤΕΧΟΝΤΩΝ ΣΤΗ ΔΟΚΙΜΗ Υγιείς εθελοντές Ασθενείς Ειδικές ευάλωτες πληθυσμιακές ομάδες Γυναίκες που είναι ικανές να κυοφορήσουν αλλά δεν κάνουν χρήση αντισύλληψης Γυναίκες που είναι ικανές να κυοφορήσουν αλλά κάνουν χρήση αντισύλληψης Έγκυες γυναίκες Γυναίκες σε γαλουχία Καταστάσεις έκτακτης ανάγκης Συμμετέχοντες ανίκανοι να συναινέσουν προσωπικά Εάν ναι, προσδιορίστε: Άλλο: Εάν ναι, προσδιορίστε: ΣΤ.4 ΣΤ.4.1 ΣΤ.4.2 ΣΤ.4.2.1 ΣΤ.4.2.2 ΠΡΟΓΡΑΜΜΑΤΙΣΜΕΝΟΣ ΑΡΙΘΜΟΣ ΣΥΜΜΕΤΕΧΟΝΤΩΝ : Στο Κράτος Μέλος: Για πολυεθνική δοκιμή: Στην ΕΟΚ Σε ολόκληρη την κλινική δοκιμή: ΣΤ.5 ΣΧΕΔΙΑ ΓΙΑ ΘΕΡΑΠΕΙΑ Ή ΠΕΡΙΘΑΛΨΗ ΜΕΤΑ ΤΟ ΠΕΡΑΣ ΤΗΣ ΣΥΜΜΕΤΟΧΗΣ ΕΝΟΣ/ΜΙΑΣ ΣΥΜΜΕΤΕΧΟΝΤΑ/ΣΥΜΜΕΤΕΧΟΥΣΑΣ ΣΤΗ ΔΟΚΙΜΗ. Παρακαλώ προσδιορίστε (ελεύθερο κείμενο): ( ) ( ( ) ) Ζ. ΠΡΟΤΕΙΝΟΜΕΝΑ ΚΕΝΤΡΑ ΔΙΕΞΑΓΩΓΗΣ/ΕΡΕΥΝΗΤΕΣ ΤΗΣ ΚΛΙΝΙΚΗΣ ΔΟΚΙΜΗΣ ΣΤΟ ΚΡΑΤΟΣ ΜΕΛΟΣ ΠΟΥ ΑΦΟΡΑ Η ΠΑΡΟΥΣΑ ΑΙΤΗΣΗ Ζ.1 Ζ.1.1 Ζ.1.2 Ζ.1.3 Ζ.1.4 ΕΡΕΥΝΗΤΗΣ ΣΥΝΤΟΝΙΣΜΟΥ (για πολυκεντρική δοκιμή) και κύριος ερευνητής (για δοκιμή σε ένα μόνο κέντρο) Όνομα Μεσαίο όνομα, εάν υπάρχει Επώνυμο Τίτλος σπουδών (Ιατρός……….) οποίων η εισαγωγή έχει εγκριθεί είναι αυτός που προσδιορίζεται στο εγκεκριμένο πρωτόκολλο ή σε μεταγενέστερες εγκεκριμένες τροποποιήσεις του. Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 14 από 25 ΔΦΜΕ-E.4200-4/2 Ζ.1.5 Ζ.1.5.1 Ζ.1.5.2 Ζ.1.5.3 Ζ.1.5.4 Ζ.1.5.5 Ζ.1.5.6 Ζ.1.6 Ζ.1.7 Ζ.1.8 Διεύθυνση εργασίας: Όνομα ιδρύματος Τμήμα Ιδρύματος Οδός Πόλη Ζ.2 ΚΥΡΙΟΣ ΕΡΕΥΝΗΤΗΣ (για πολυκεντρική δοκιμή - όπου απαιτείται, χρησιμοποιήστε επιπλέον έντυπα) Όνομα Μεσαίο όνομα, εάν υπάρχει Επώνυμο Τίτλος σπουδών (Ιατρός……….) Διεύθυνση εργασίας: Οδός Πόλη Ταχυδρομικός Κώδικας Χώρα Τηλέφωνο: Fax: E-mail: Ζ.2.1 Ζ.2.2 Ζ.2.3 Ζ.2.4 Ζ.2.5 Z.2.5.1 Z.2.5.2 Z.2.5.3 Z.2.5.4 Z.2.6 Z.2.7 Z.2.8 Ζ.3 Z.3.1 Ζ.3.2 Ζ.3.3 Ζ.3.3.1 Ζ.3.3.2 Ζ.3.3.3 Z.3.4 Z.3.4.1 Z.3.4.2 Z.3.4.3 Z.3.4.4 Z.3.5 Z.3.6 Z.3.7 Z.3.8 1.1.3 Ταχυδρομικός Κώδικας Χώρα Τηλέφωνο: Fax: E-mail: ΚΕΝΤΡΙΚΕΣ ΤΕΧΝΙΚΕΣ ΕΓΚΑΤΑΣΤΑΣΕΙΣ ΠΟΥ ΘΑ ΧΡΗΣΙΜΟΠΟΙΗΘΟΥΝ ΓΙΑ ΤΗ ΔΙΕΝΕΡΓΕΙΑ ΤΗΣ ΔΟΚΙΜΗΣ Εργαστήριο ή άλλη τεχνική εγκατάσταση, όπου συγκεντρώνονται τα στοιχεία από τη μέτρηση ή εκτίμηση των κύριων κριτηρίων αξιολόγησης (επαναλάβετε όπως απαιτείται όταν πρόκειται για πολλούς οργανισμούς) Οργανισμός: Τμήμα: Ονοματεπώνυμο υπευθύνου επικοινωνίας: Όνομα Μεσαίο Όνομα Επώνυμο Διεύθυνση: Οδός Πόλη Ταχυδρομικός Κώδικας Χώρα Τηλέφωνο: Fax: E-mail: Αρμοδιότητες που ανατέθηκαν με υπεργολαβία: Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 15 από 25 ΔΦΜΕ-E.4200-4/2 Ζ.4 Z.4.1 Z.4.2 Z.4.2.1 Z.4.2.2 Z.4.2.3 Z.4.3 Z.4.3.1 Z.4.3.2 Z.4.3.3 Z.4.3.4 Z.4.4 Z.4.5 Z.4.6 Z.4.7 ΔΙΚΤΥΑ ΠΟΥ ΕΝΕΧΟΝΤΑΙ ΣΤΗΝ ΔΟΚΙΜΗ) (π.χ. Παιδιατρικά Δίκτυα που ενέχονται στην δοκιμή) Οργανισμός: Ονοματεπώνυμο υπευθύνου επικοινωνίας: Όνομα Μεσαίο Όνομα Επώνυμο Διεύθυνση: Οδός Πόλη Ταχυδρομικός Κώδικας Χώρα Τηλέφωνο: Fax: E-mail: Αρμοδιότητες που ανατέθηκαν στο δίκτυο: Ζ.5 ΟΡΓΑΝΙΣΜΟΙ ΣΤΟΥΣ ΟΠΟΙΟΥΣ Ο ΧΟΡΗΓΟΣ ΕΧΕΙ ΜΕΤΑΦΕΡΕΙ ΚΑΘΗΚΟΝΤΑ ΚΑΙ ΑΡΜΟΔΙΟΤΗΤΕΣ ΠΟΥ ΣΧΕΤΙΖΟΝΤΑΙ ΜΕ ΤΗ ΔΟΚΙΜΗ (επαναλάβετε όπως απαιτείται όταν πρόκειται για πολλούς οργανισμούς) ναι όχι Ζ.5.1 Έχει μεταβιβάσει ο χορηγός σημαντικό μέρος ή το σύνολο των καθηκόντων και αρμοδιοτήτων του που σχετίζονται με τη δοκιμή σε άλλο οργανισμό ή σε τρίτο μέρος; Επαναλάβετε όπως απαιτείται όταν πρόκειται για πολλούς οργανισμούς: Ζ.5.1.1 Οργανισμός: Ζ.5.1.2 Τμήμα: Ζ.5.1.3 Ονοματεπώνυμο υπευθύνου επικοινωνίας: Ζ.5.1.3.1 Όνομα Ζ.5.1.3.2 Μεσαίο Όνομα Ζ.5.1.3.3 Επώνυμο Ζ.5.1.4 Διεύθυνση: Ζ.5.1.4.1 Οδός Ζ.5.1.4.2 Πόλη Ζ.5.1.4.3 Ταχυδρομικός Κώδικας Ζ.5.1.4.4 Χώρα Ζ.5.1.5 Τηλέφωνο Ζ.5.1.6 Fax Ζ.5.1.7 E-mail Ζ.5.1.8 Ζ.5.1.9 Ζ.5.1.10 Ζ.5.1.11 Ζ.5.1.12 Ζ.5.1.13 Ζ.4.1.14 Ζ.5.1.15 Ζ.5.1.16 Ζ.5.1.17 Ζ.5.1.18 Όλα τα καθήκοντα του χορηγού Επιτήρηση (monitoring) Εγκριτικές δραστηριότητες (π.χ. προετοιμασία αιτήσεων για ΕΟΦ/ΕΕΔ) Στρατολόγηση ερευνητών IVRS34– τυχαιοποίηση θεραπείας Διαχείριση δεδομένων Συλλογή ηλεκτρονικών δεδομένων (E-data capture) Αναφορά SUSAR Επιθεώρηση διασφάλισης ποιότητας Στατιστική ανάλυση Συγγραφή ιατρικών κειμένων (medical writing) ναι ναι ναι ναι ναι ναι ναι ναι ναι ναι ναι όχι όχι όχι όχι όχι όχι όχι όχι όχι όχι όχι Ζ.5.1.19 Ζ.5.1.19.1 Άλλα καθήκοντα που έχουν ανατέθηκαν σε τρίτους Εάν ναι, προσδιορίστε: ναι όχι Η. ΑΡΜΟΔΙΑ ΑΡΧΗ / ΕΠΙΤΡΟΠΗ ΔΕΟΝΤΟΛΟΓΙΑΣ ΣΤΟ ΚΡΑΤΟΣ ΜΕΛΟΣ ΠΟΥ ΑΦΟΡΑ Η ΠΑΡΟΥΣΑ ΑΙΤΗΣΗ 34 Interactive Voice Response System: συνήθως χρησιμοποιείται για την τυχαιοποίηση της θεραπείας και τον έλεγχο της διακίνησης του αποθέματος του φαρμάκου. Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 16 από 25 ΔΦΜΕ-E.4200-4/2 Η.1 ΤΥΠΟΣ ΤΗΣ ΑΙΤΗΣΗΣ Εάν η παρούσα αίτηση απευθύνεται στην αρμόδια αρχή, σημειώστε στο τετραγωνίδιο της Επιτροπής Δεοντολογίας και παρέχετε πληροφορίες σχετικά με την ενεχόμενη Επιτροπή Δεοντολογίας. Εάν η παρούσα αίτηση απευθύνεται στην Επιτροπή Δεοντολογίας, σημειώστε στο τετραγωνίδιο της αρμόδιας αρχής και παρέχετε πληροφορίες σχετικά με την ενεχόμενη αρμόδια αρχή. Η.1.1 Αρμόδια αρχή (ΕΟΦ) Η.1.2 Επιτροπή δεοντολογίας (ΕΕΔ) Η.2 Η.2.1 Η.2.2 Η.2.2.1 Η.2.2.2 Η.2.2.3 Η.2.2.4 Η.2.3 ΠΛΗΡΟΦΟΡΙΕΣ ΓΙΑ ΤΗΝ ΑΡΜΟΔΙΑ ΑΡΧΗ / ΕΠΙΤΡΟΠΗ ΔΕΟΝΤΟΛΟΓΙΑΣ Ονομασία: Διεύθυνση Οδός Πόλη Ταχυδρομικός Κώδικας Χώρα Ημερομηνία υποβολής: Η.3 ΧΟΡΗΓΗΣΗ ΑΔΕΙΑΣ/ΓΝΩΜΟΔΟΤΗΣΗΣ: Η.3.1 Υπό αίτηση Η.3.2 Εκκρεμεί Η.3.3 Έχει δοθεί Εάν «έχει δοθεί», προσδιορίστε: Η.3.3.1 Ημερομηνία χορήγησης αδείας / γνωμοδότησης: Η.3.3.2 Αποδεκτή χορήγηση αδείας / θετική γνωμοδότηση Η.3.3.3 Μη αποδεκτή / μη θετική Σε περίπτωση μη αποδεκτής / αρνητικής, αναφέρετε: Η.3.3.3.1 Τους λόγους Η.3.3.3.2 Την ενδεχόμενη ημερομηνία νέας υποβολής: Θ. ΥΠΟΓΡΑΦΗ ΤΟΥ ΑΙΤΟΥΝΤΟΣ ΣΤΟ ΚΡΑΤΟΣ ΜΕΛΟΣ Θ.1 Διά της παρούσης βεβαιώ ότι / βεβαιώ εκ μέρους του χορηγού ότι (διαγράψτε κατά περίπτωση): Οι πληροφορίες που διατίθενται είναι πλήρεις, Τα επισυναπτόμενα έγγραφα περιέχουν επακριβώς τις διαθέσιμες πληροφορίες , Η κλινική δοκιμή θα διενεργηθεί σύμφωνα με το πρωτόκολλο, Η διεξαγωγή της κλινικής δοκιμής, οι αναφορές για τα SUSARs και οι πληροφορίες των αποτελεσμάτων, θα γίνονται σύμφωνα με την εφαρμοζόμενη νομοθεσία. Θ.2 Θ.2.1 Θ.2.2 Θ.2.3 Ο ΥΠΟΒΑΛΛΩΝ ΤΗΝ ΑΙΤΗΣΗ ΠΡΟΣ ΤΗΝ ΑΡΜΟΔΙΑ ΑΡΧΗ (ΕΟΦ) (όπως αναφέρεται στο τμήμα Γ.1): Ημερομηνία: Υπογραφή35: Ονοματεπώνυμο: Θ.3 Ο ΥΠΟΒΑΛΛΩΝ ΤΗΝ ΑΙΤΗΣΗ ΠΡΟΣ ΤΗΝ ΕΠΙΤΡΟΠΗ ΔΕΟΝΤΟΛΟΓΙΑΣ (ΕΕΔ) (όπως αναφέρεται στο τμήμα Γ.2): Ημερομηνία: Υπογραφή36: Ονοματεπώνυμο: Θ.3.1 Θ.3.2 Θ.3.3 Παράρτημα 2: Αίτηση ουσιώδους τροποποίησης (Ανατρέξτε στο κεφάλαιο 3.7.b του εγγράφου ¨Λεπτομερείς Οδηγίες σχετικά με την αίτηση προς τις αρμόδιες αρχές για τη χορήγηση άδειας με σκοπό τη διενέργεια κλινικής δοκιμής, όσον αφορά 35 36 Στην αίτηση που απευθύνεται προς τον ΕΟΦ, απαιτείται να υπογράψει μόνο ο αιτών προς τον ΕΟΦ. Στην αίτηση που απευθύνεται προς την ΕΕΔ, απαιτείται να υπογράψει μόνο ο αιτών προς την ΕΕΔ. Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 17 από 25 ΔΦΜΕ-E.4200-4/2 φαρμακευτικό προϊόν που προορίζεται για ανθρώπινη χρήση, τη γνωστοποίηση ουσιωδών τροποποιήσεων και τη δήλωση περάτωσης της κλινικής δοκιμής37¨ ) ΓΝΩΣΤΟΠΟΙΗΣΗ ΟΥΣΙΩΔΟΥΣ ΤΡΟΠΟΠΟΙΗΣΗΣ ΚΛΙΝΙΚΗΣ ΔΟΚΙΜΗΣ ΦΑΡΜΑΚΕΥΤΙΚΟΥ ΠΡΟΪΟΝΤΟΣ ΓΙΑ ΑΝΘΡΩΠΙΝΗ ΧΡΗΣΗ ΠΡΟΣ ΤΟΝ ΕΘΝΙΚΟ ΟΡΓΑΝΙΣΜΟ ΦΑΡΜΑΚΩΝ (ΕΟΦ) ΚΑΙ ΓΙΑ ΓΝΩΜΟΔΟΤΗΣΗ ΤΗΣ ΕΘΝΙΚΗΣ ΕΠΙΤΡΟΠΗΣ ΔΕΟΝΤΟΛΟΓΙΑΣ (ΕΕΔ) Συμπληρώνεται από την υπηρεσία: Ημερομηνία παραλαβής της αίτησης: Λόγοι μη αποδοχής/ αρνητικής γνώμης: Ημερομηνία: Ημερομηνία έναρξης της διαδικασίας Στον ΕΟΦ: Χορήγηση αδείας / θετική γνώμη: Ημερομηνία: Αριθμός καταχώρησης πρωτοκόλλου ΕΟΦ για τη τροποποίηση: Αριθμός καταχώρησης πρωτοκόλλου ΕΕΔ για τη τροποποίηση: Ανάκληση της αίτησης τροποποίησης Ημερομηνία: Συμπληρώνεται από τον αιτούντα: Το παρόν έντυπο χρησιμοποιείται τόσο για αίτηση προς τον ΕΟΦ για έγκριση ουσιώδους τροποποίησης όσο και προς την ΕΕΔ για τη γνωμοδότηση της για μια ουσιώδη τροποποίηση. Υποδείξτε ανάλογα το σκοπό της αίτησης στο Τμήμα Α. Α. ΤΥΠΟΣ ΚΟΙΝΟΠΟΙΗΣΗΣ Α.1 Α.2 Α.3 Κράτος μέλος στο οποίο υποβάλλεται η ουσιώδης τροποποίηση: Κοινοποίηση για χορήγηση αδείας προς τον ΕΟΦ: Κοινοποίηση για γνωμοδότηση της ΕΕΔ: Β. ΣΤΟΙΧΕΙΑ ΚΛΙΝΙΚΗΣ ΔΟΚΙΜΗΣ (Όταν η τροποποίηση αφορά περισσότερες από μία κλινικές δοκιμές, χρησιμοποιήστε τον απαραίτητο αριθμό εντύπων.) ναι όχι Β.1 Η ουσιώδης τροποποίηση αφορά σε αρκετές κλινικές δοκιμές με το ίδιο 38 ΥΕΦΠ ; Εάν ναι επαναλάβετε το τμήμα όπως απαιτείται. Β.1.1 Β.2 Β.3 Β.4 Αριθμός EudraCT: Πλήρης τίτλος της κλινικής δοκιμής: Κωδικός αριθμός, έκδοση και ημερομηνία πρωτοκόλλου του χορηγού: Γ. ΣΤΟΙΧΕΙΑ ΧΟΡΗΓΟΥ ΥΠΕΥΘΥΝΟΥ ΓΙΑ ΤΗΝ ΑΙΤΗΣΗ Γ.1 Γ.1.1 Γ.1.2 Γ.1.3 Γ.1.4 Γ.1.5 Γ.1.6 37 38 Χορηγός Οργανισμός: Ονοματεπώνυμο υπεύθυνου επικοινωνίας: Διεύθυνση: Αριθμός τηλεφώνου: Αριθμός fax: e-mail: OJ, C82, 30.3.2010, σελ.1; στο εφ’ εξής αναφερόμενο ως ‘Λεπτομερείς Οδηγίες CT-1’. Ανατρέξτε στο κεφάλαιο 3.7 του CT-1 Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 18 από 25 ΔΦΜΕ-E.4200-4/2 Γ.2 Γ.2.1 Γ.2.2 Γ.2.3 Γ.2.4 Γ.2.5 Γ.2.6 Νόμιμος εκπρόσωπος39 του χορηγού στην Ε.Ε για το σκοπό της εν λόγω κλινικής δοκιμής (εάν είναι διαφορετικός από τον χορηγό) Οργανισμός: Ονοματεπώνυμο υπεύθυνου επικοινωνίας: Διεύθυνση: Αριθμός τηλεφώνου: Αριθμός fax: e-mail: Δ. ΣΤΟΙΧΕΙΑ ΑΙΤΟΥΝΤΟΣ, (σημειώστε στο κατάλληλο τετραγωνίδιο) Δ.1 Δ.1.1 Δ.1.2 Δ.1.3 Δ.1.4 Δ.1.4.1 Δ.1.4.2 Δ.1.4.3 Δ.1.4.4 Δ.1.4.5 Δ.1.4.6 Αίτηση προς τον ΕΟΦ Χορηγός Νόμιμος εκπρόσωπος χορηγού Πρόσωπο ή οργανισμός εξουσιοδοτημένος από το χορηγό να υποβάλει την αίτηση Συμπληρώστε παρακάτω: Οργανισμός: Ονοματεπώνυμο υπεύθυνου επικοινωνίας: Διεύθυνση: Αριθμός τηλεφώνου: Αριθμός fax: e-mail: Δ.2 Δ.2.1 Δ.2.2 Δ.2.3 Δ.2.4 Αίτηση προς την ΕΕΔ Χορηγός Νόμιμος εκπρόσωπος χορηγού Πρόσωπο ή οργανισμός εξουσιοδοτημένος από το χορηγό να υποβάλει την αίτηση Ερευνητής υπεύθυνος για την αίτηση εάν υπάρχει:40 Ερευνητής συντονισμού (για πολυκεντρική κλινική δοκιμή) Κύριος ερευνητής (για μονοκεντρική κλινική δοκιμή) Συμπληρώστε παρακάτω: Οργανισμός: Ονοματεπώνυμο υπεύθυνου επικοινωνίας: Διεύθυνση: Αριθμός τηλεφώνου: Αριθμός fax: e-mail: Δ.2.5 Δ.2.5.1 Δ.2.5.2 Δ.2.5.3 Δ.2.5.4 Δ.2.5.5 Δ.2.5.6 39 40 Όπως αναφέρεται στο Άρθρο 19 της Οδηγίας 2001/20/ΕΚ. Σύμφωνα με την εθνική νομοθεσία. Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 19 από 25 ΔΦΜΕ-E.4200-4/2 Ε. ΠΡΟΣΔΙΟΡΙΣΜΟΣ ΟΥΣΙΩΔΟΥΣ ΤΡΟΠΟΠΟΙΗΣΗΣ Ε.1 Κωδικός αριθμός, έκδοση και ημερομηνία ουσιώδους τροποποίησης του χορηγού για τη σχετιζόμενη κλινική δοκιμή: ( ) Ε.2 Ε.2.1 Ε.2.2 Ε.2.3 Ε.2.3.1 Ε.2.4 Ε.2.4.1 Ε.2.5 Ε.2.6 Ε.2.7 Ε.3 Ε.3.1 Ε.3.2 Ε.3.3 Ε.3.4 Ε.3.5 Ε.3.6 Ε.3.7 Ε.3.7.1 Ε.3.8 Ε.3.8.1 Ε.4 Ε.4.1 Ε.4.2 Ε.4.3 Ε.4.4 Ε.4.5 Τύπος ουσιώδους τροποποίησης Τροποποίηση πληροφοριών στο έντυπο της αίτησης Τροποποίηση επί του πρωτοκόλλου Τροποποίηση σε άλλα επισυναπτόμενα έγγραφα στην αρχική αίτηση Εάν ναι προσδιορίστε: Τροποποίηση σε άλλα έγγραφα ή στοιχεία Εάν ναι προσδιορίστε: Η τροποποίηση αφορά κυρίως έκτακτα μέτρα ασφαλείας που έχουν ήδη εφαρμοστεί41 Η τροποποίηση αφορά την κοινοποίηση μιας προσωρινής διακοπής της κλινικής δοκιμής42 Η τροποποίηση αφορά την αίτηση επανέναρξης της κλινικής δοκιμής43 Λόγοι ουσιώδους τροποποίησης Αλλαγές στην ασφάλεια ή ακεραιότητα των συμμετεχόντων στη κλινική δοκιμή: Αλλαγές στην ερμηνεία των επιστημονικών εγγράφων / αξία της κλινικής δοκιμής Αλλαγές στην ποιότητα του/των υπό έρευνα φαρμακευτικού(ων) προϊόντος(ων) (ΥΕΦΠ) Αλλαγές στη διεξαγωγή ή διαχείριση της κλινικής δοκιμής Αλλαγή ή προσθήκη κύριου ερευνητή(ών), συντονίζοντος ερευνητή: Αλλαγή / προσθήκη κέντρου(ων) Άλλη αλλαγή: Εάν ναι, προσδιορίστε: Άλλη περίπτωση: Εάν ναι, προσδιορίστε: Πληροφορίες σχετικά με την προσωρινή διακοπή της κλινικής δοκιμής44 Ημερομηνία της προσωρινής διακοπής (ΧΧΧΧ/ΜΜ/HH) Έχει γίνει διακοπή της προσέλκυσης Έχει γίνει διακοπή της θεραπείας Αριθμός ασθενών που εξακολουθούν να υποβάλλονται σε αγωγή τη στιγμή της προσωρινής διακοπής στο ενεχόμενο Κράτος Μέλος Περιγράψτε εν συντομία (ελεύθερο κείμενο): - Αιτιολόγηση για μια προσωρινή διακοπή της κλινικής δοκιμής - Την προτεινόμενη διαχείριση των ασθενών που εξακολουθούν να υποβάλλονται σε αγωγή τη στιγμή της διακοπής (ελεύθερο κείμενο) - Τις συνέπειες της προσωρινής διακοπής στην αξιολόγηση των αποτελεσμάτων και στη συνολική αποτίμηση της σχέσης κινδύνου/οφέλους του υπό έρευνα φαρμακευτικού προϊόντος (ελεύθερο κείμενο) ναι ναι ναι όχι όχι όχι ναι όχι ναι όχι ναι όχι ναι όχι ναι όχι ναι όχι ναι όχι ναι ναι ναι ναι ναι όχι όχι όχι όχι όχι ναι όχι ναι ναι όχι όχι 41 Ανατρέξτε στο κεφάλαιο 3.9 του CT-1 Ανατρέξτε στο κεφάλαιο 3.10 του CT-1 43 Ανατρέξτε στο κεφάλαιο 3.10 του CT-1 44 Ανατρέξτε στο κεφάλαιο 3.10 του CT-1 42 Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 20 από 25 ΔΦΜΕ-E.4200-4/2 ΣΤ. ΠΕΡΙΓΡΑΦΗ ΤΩΝ ΟΥΣΙΩΔΩΝ ΤΡΟΠΟΠΟΙΗΣΕΩΝ45(ελεύθερο κείμενο): Προηγούμενη και νέα διατύπωση των αλλαγών 45 Νέα διατύπωση Σχόλια/Επεξηγήσεις/Αιτιολόγησ η ουσιωδών τροποποιήσεων Ανατρέξτε στο κεφάλαιο 3.7(γ) του CT-1. O χορηγός μπορεί να καταθέσει αυτό το έγγραφο σε ξεχωριστεί σελίδα. Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 21 από 25 ΔΦΜΕ-E.4200-4/2 Ζ. ΑΛΛΑΓΕΣ ΣΤΟ ΚΕΝΤΡΟ(Α)/ΣΤΟΝ(ΟΥΣ)ΕΡΕΥΝΗΤΗ(ΕΣ) ΤΗΣ ΚΛΙΝΙΚΗΣ ΔΟΚΙΜΗΣ ΣΤΟ ΚΡΑΤΟΣ ΜΕΛΟΣ ΠΟΥ ΑΦΟΡΑ Η ΠΑΡΟΥΣΑ ΤΡΟΠΟΠΟΙΗΣΗ Ζ.1 Ζ.1.1 Ζ.1.1.1 Ζ.1.1.1.1 Ζ.1.1.1.2 Ζ.1.1.1.3 Ζ.1.1.1.4 Ζ.1.1.1.5 Ζ.1.2 Ζ.1.2.1 Ζ.1.2.1.1 Ζ.1.2.1.2 Ζ.1.2.1.3 Ζ.1.2.1.4 Ζ.1.2.1.5 Ζ.1.3 Ζ.1.3.1 Ζ.1.3.2 Ζ.1.3.3 Ζ.1.3.4 Ζ.1.3.5 Ζ.1.3.6 Ζ.1.4 Ζ.1.4.1 Ζ.1.4.2 Ζ.1.4.3 Ζ.1.4.4 Ζ.1.4.5 Ζ.1.4.6 Τύπος της αλλαγής Προσθήκη νέου κέντρου Κύριος ερευνητής (δώστε λεπτομέρειες παρακάτω) Όνομα Μεσαίο όνομα, εάν υπάρχει Επώνυμο Τίτλος σπουδών (Ιατρός……….) Διεύθυνση επαγγέλματος Αφαίρεση εγκεκριμένου κέντρου Κύριος ερευνητής (δώστε λεπτομέρειες παρακάτω) Όνομα Μεσαίο όνομα, εάν υπάρχει Επώνυμο Τίτλος σπουδών (Ιατρός……….) Διεύθυνση επαγγέλματος Αλλαγή του ερευνητή συντονισμού (δώστε λεπτομέρειες παρακάτω για το νέο ερευνητή συντονισμού) Όνομα Μεσαίο όνομα Επώνυμο Τίτλος σπουδών (Ιατρός……….) Διεύθυνση επαγγέλματος Αναφέρετε το όνομα του προηγούμενου ερευνητή συντονισμού: Αλλαγή του κύριου ερευνητή σε εγκεκριμένο κέντρο (δώστε λεπτομέρειες παρακάτω για το νέο κύριο ερευνητή) Όνομα Μεσαίο όνομα Επώνυμο Τίτλος σπουδών (Ιατρός……….) Διεύθυνση επαγγέλματος Αναφέρετε το όνομα του προηγούμενου κύριου ερευνητή: Η. ΑΛΛΑΓΗ ΤΩΝ ΟΔΗΓΙΩΝ ΠΡΟΣ ΤΟΝ ΕΟΦ ΓΙΑ ΕΠΑΝΕΝΗΜΕΡΩΣΗ ΤΟΥ ΧΟΡΗΓΟΥ Η.1 Η.2 Αλλαγή των ηλεκτρονικών (e-mail) διευθύνσεων για επανενημέρωση του χορηγού* Αλλαγή στην αίτηση για λήψη .xml αντίγραφου των δεδομένων της αίτησης για ναι κλινική δοκιμή (CTA); Η.2.1 Επιθυμείτε ένα .xml αντίγραφο των δεδομένων της CTA όπως αποθηκεύτηκε στο ναι EudraCT; Η.2.1.1 Εάν ναι, παραθέστε την/τις ηλεκτρονική/ές διεύθυνση/σεις (e-mail) στις οποίες θα πρέπει να αποσταλεί το αντίγραφο (έως 5 διευθύνσεις): Η.2.2 Επιθυμείτε την παραλαβή μέσω σύνδεσης(ων) προστατευμένης(ων) με κωδικό;46 ναι Εάν απαντήσετε όχι στην ερώτηση Θ.2.2 το .xml αρχείο θα μεταδοθεί μέσω σύνδεσης(ων) ηλεκτρονικού ταχυδρομείου (e-mail links) μειωμένης ασφάλειας Η.2.3 Θέλετε να πάψει η αποστολή μηνυμάτων προς μια ηλεκτρονική διεύθυνση (eναι mail) προς την οποία είχατε προηγουμένως ζητήσει να αποστέλλονται; Η.2.3.1 Εάν ναι δώστε την/τις ηλεκτρονική/ές διεύθυνση/διευθύνσεις οι οποίες δεν θα πρέπει να πλέον να ενημερώνονται 46 όχι όχι όχι όχι Απαιτείται ένας λογαριασμός EudraLink (βλέπε www.eudract.emea.eu.int για λεπτομέρειες) Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 22 από 25 ΔΦΜΕ-E.4200-4/2 (* Τίθεται σε εφαρμογή μόνο από τη στιγμή κατά την οποία το αίτημα επεξεργάζεται στο EudraCT) Θ. ΚΑΤΑΛΟΓΟΣ ΤΩΝ ΕΓΓΡΑΦΩΝ ΠΟΥ ΕΠΙΣΥΝΑΠΤΟΝΤΑΙ ΣΤΟ ΕΝΤΥΠΟ ΤΗΣ ΑΙΤΗΣΗΣ(Ανατρέξτε στο κεφάλαιο 3.10 του CT-1): Υποβάλλετε μόνο σχετικά έγγραφα και/ή εφόσον συντρέχει περίπτωση, προσδιορίστε με σαφή τρόπο τα έγγραφα που έχουν ήδη υποβληθεί. Προσδιορίστε με σαφή τρόπο τυχόν αλλαγές σε μεμονωμένες σελίδες και υποβάλλετε μαζί τα παλιά και τα νέα κείμενα. Σημειώστε στο ανάλογο τετραγωνίδιο(-α). Θ.1 Θ.2 Θ.3 Θ.4 Θ.5 Θ.6 Συνοδευτική επιστολή Απόσπασμα από το τροποποιημένο έγγραφο σύμφωνα με το κεφάλαιο 3.7 του CT-1 (εάν δεν περιλαμβάνεται ήδη στο Κεφάλαιο ΣΤ της παρούσας αίτησης) Νέα έκδοση εγγράφου47 Υποστηρικτικά στοιχεία Τροποποιημένο .XML αρχείο και αντίγραφο της αρχικής αίτησης με υπογραμμισμένα τα τροποποιημένα στοιχεία Σχόλιο σε οποιαδήποτε νεότερη επιστημονική άποψη που αφορά την τροποποίηση εάν υπάρχει: Ι. ΥΠΟΓΡΑΦΗ ΤΟΥ ΑΙΤΟΥΝΤΟΣ ΣΤΟ ΚΡΑΤΟΣ ΜΕΛΟΣ: Ι.1 Δια της παρούσης βεβαιώ ότι / βεβαιώ εκ μέρους του χορηγού ότι: (διαγράψτε ότι δεν ισχύει) Οι πληροφορίες που περιέχονται στην παρούσα αίτηση είναι ακριβείς, Η κλινική δοκιμή θα διενεργηθεί σύμφωνα με το πρωτόκολλο, τους εθνικούς κανονισμούς και τις αρχές της ορθής κλινικής πρακτικής και Θεωρώ εύλογη τη διενέργεια της προτεινόμενης τροποποίησης Ι.2 Ο ΥΠΟΒΑΛΛΩΝ ΤΗΝ ΑΙΤΗΣΗ ΠΡΟΣ ΤΗΝ ΑΡΜΟΔΙΑ ΑΡΧΗ (ΕΟΦ) (όπως αναφέρεται στο τμήμα Γ.1) Υπογραφή48: Ονοματεπώνυμο: Ημερομηνία: Ι.2.1 Ι.2.2 Ι.2.3 Ι.3 Ι.3.1 Ι.3.2 Ι.3.3 Ο ΥΠΟΒΑΛΛΩΝ ΤΗΝ ΑΙΤΗΣΗ ΠΡΟΣ ΤΗΝ ΔΕΟΝΤΟΛΟΓΙΑΣ (ΕΕΔ) (όπως αναφέρεται στο τμήμα Γ.2) Υπογραφή49: Ονοματεπώνυμο: Ημερομηνία: ΕΘΝΙΚΗ ΕΠΙΤΡΟΠΗ Παράρτημα 3: Δήλωση περάτωσης κλινικής δοκιμής (Ανατρέξτε στο κεφάλαιο 4.2.1 του εγγράφου ¨Λεπτομερείς Οδηγίες σχετικά με την αίτηση προς τις αρμόδιες αρχές για τη χορήγηση άδειας με σκοπό τη διενέργεια κλινικής δοκιμής, όσον αφορά φαρμακευτικό προϊόν που προορίζεται για ανθρώπινη χρήση, τη γνωστοποίηση ουσιωδών τροποποιήσεων και τη δήλωση περάτωσης της κλινικής δοκιμής50¨) Ανατρέξτε στο κεφάλαιο 3.7(γ) του CT-1. Στην αίτηση που απευθύνεται προς τον ΕΟΦ, απαιτείται να υπογράψει μόνο ο αιτών προς τον ΕΟΦ. 49 Στην αίτηση που απευθύνεται προς την ΕΕΔ, απαιτείται να υπογράψει μόνο ο αιτών προς την ΕΕΔ. 50 OJ, C82, 30.3.2010, σελ. 1: στο εφ’ εξής αναφερόμενο ως ‘Λεπτομερείς Οδηγίες CT-1’. 47 48 Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 23 από 25 ΔΦΜΕ-E.4200-4/2 ΚΟΙΝΟΠΟΙΗΣΗ ΠΡΟΣ ΤΟΝ ΕΟΦ ΚΑΙ ΤΗΝ ΕΘΝΙΚΗ ΕΠΙΤΡΟΠΗ ΔΕΟΝΤΟΛΟΓΙΑΣ (ΕΕΔ) ΔΗΛΩΣΗΣ ΠΕΡΑΤΩΣΗΣ ΚΛΙΝΙΚΗΣ ΔΟΚΙΜΗΣ ΦΑΡΜΑΚΕΥΤΙΚΟΥ ΠΡΟΪΟΝΤΟΣ ΓΙΑ ΑΝΘΡΩΠΙΝΗ ΧΡΗΣΗ Συμπληρώνεται από την υπηρεσία: Ημερομηνία παραλαβής της αίτησης: Αριθμός πρωτοκόλλου ΕΟΦ για τη περάτωση της κλινικής δοκιμής: Αριθμός πρωτοκόλλου ΕΕΔ για τη περάτωση της κλινικής δοκιμής: Συμπληρώνεται από τον αιτούντα: Α. ΚΡΑΤΟΣ ΜΕΛΟΣ ΣΤΟ ΟΠΟΙΟ ΓΙΝΕΤΑΙ Η ΔΗΛΩΣΗ: Β. ΣΤΟΙΧΕΙΑ ΚΛΙΝΙΚΗΣ ΔΟΚΙΜΗΣ Β.1 Β.2 Β.3 Αριθμός EudraCT: Κωδικός αριθμός πρωτοκόλλου του χορηγού: Πλήρης τίτλος της κλινικής δοκιμής : (..) (..) Γ. ΣΤΟΙΧΕΙΑ ΑΙΤΟΥΝΤΟΣ (σημειώστε στο κατάλληλο τετραγωνίδιο) Γ.1 Γ.1.1 Γ.1.2 Γ.1.3 Γ.1.4 Γ.1.4.1 Γ.1.4.2 Γ.1.4.3 Γ.1.4.4 Γ.1.4.5 Γ.1.4.6 ΔΗΛΩΣΗ ΠΡΟΣ ΤΟΝ ΕΟΦ Χορηγός Νόμιμος εκπρόσωπος χορηγού Πρόσωπο ή οργανισμός εξουσιοδοτημένος από το χορηγό να υποβάλει την αίτηση Συμπληρώστε παρακάτω: Οργανισμός: Ονοματεπώνυμο υπεύθυνου επικοινωνίας: Διεύθυνση: Αριθμός τηλεφώνου: Αριθμός fax: e-mail: Γ.2 Γ.2.1 Γ.2.2 Γ.2.3 Γ.2.4 ΔΗΛΩΣΗ ΠΡΟΣ ΤΗΝ ΕΕΔ Χορηγός Νόμιμος εκπρόσωπος χορηγού Πρόσωπο ή οργανισμός εξουσιοδοτημένος από το χορηγό να υποβάλει την αίτηση Ερευνητής υπεύθυνος για την αίτηση εάν υπάρχει:51 Ερευνητής συντονισμού (για πολυκεντρική κλινική δοκιμή) Κύριος ερευνητής (για μονοκεντρική κλινική δοκιμή) Συμπληρώστε παρακάτω: Γ.2.5 51 Σύμφωνα με την εθνική νομοθεσία Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 24 από 25 ΔΦΜΕ-E.4200-4/2 Γ.2.5.1 Γ.2.5.2 Γ.2.5.3 Γ.2.5.4 Γ.2.5.5 Γ.2.5.6 Οργανισμός: Ονοματεπώνυμο υπεύθυνου επικοινωνίας: Διεύθυνση: Αριθμός τηλεφώνου: Αριθμός fax: e-mail: Δ. ΠΕΡΑΤΩΣΗ ΤΗΣ ΜΕΛΕΤΗΣ Δ.1 Δ.1.1 Δ.2 Δ.2.1 Δ.2.2 Δ.2.2.1 Δ.2.2.2 Δ.2.2.3 Ημερομηνία περάτωσης της πλήρους κλινικής δοκιμής σε όλες τις χώρες στις οποίες διεξήχθη ( ΧΧΧΧ/MM/HH) ναι όχι Η κλινική δοκιμή έχει περατωθεί πρόωρα52; Εάν ναι δώστε ημερομηνία ( ΧΧΧΧ/MM/HH) Περιγράψτε εν συντομία σε παράρτημα (ελεύθερο κείμενο): - Την αιτιολόγηση της πρόωρης διακοπής της κλινικής δοκιμής - Τον αριθμό των ασθενών που εξακολουθούν να υποβάλλονται σε αγωγή κατά το χρόνο της πρόωρης περάτωσης στο κράτος μέλος στο οποίο αφορά η δήλωση και η προτεινόμενη διαχείρισή τους - Τις συνέπειες της πρόωρης περάτωσης στην αξιολόγηση των αποτελεσμάτων και στη συνολική αποτίμηση της σχέσης κινδύνου/οφέλους για το υπό έρευνα φαρμακευτικό προϊόν. Ε. ΥΠΟΓΡΑΦΗ ΤΟΥ ΑΙΤΟΥΝΤΟΣ ΣΤΟ ΚΡΑΤΟΣ ΜΕΛΟΣ: Ε.1 Δια της παρούσης βεβαιώ ότι / βεβαιώ εκ μέρους του χορηγού ότι: (διαγράψτε όποιο δεν ισχύει) Οι ως άνω πληροφορίες που παρέχονται στην παρούσα δήλωση είναι ακριβείς και Θα υποβάλω τη σύνοψη της έκθεση της κλινικής μελέτης στον ΕΟΦ και στην ΕΕΔ, όταν αυτή θα είναι έτοιμη, και εντός προθεσμίας 1 έτους μετά την περάτωση της κλινικής δοκιμής σε όλες τις χώρες Ε.2 Ε.2.1 Ε.2.2 Ε.2.3 Ο ΥΠΟΒΑΛΛΩΝ ΤΗΝ ΑΙΤΗΣΗ ΠΡΟΣ ΤΗΝ ΑΡΜΟΔΙΑ ΑΡΧΗ (ΕΟΦ) (όπως αναφέρεται στο τμήμα Γ.1) Υπογραφή: Ονοματεπώνυμο: Ημερομηνία: Ε.3 Ε.3.1 Ε.3.2 Ε.3.3 Ο ΥΠΟΒΑΛΛΩΝ ΤΗΝ ΑΙΤΗΣΗ ΠΡΟΣ ΤΗΝ ΕΕΔ (όπως αναφέρεται στο τμήμα Γ.2) Υπογραφή: Ονοματεπώνυμο: Ημερομηνία: 52 Ανατρέξτε στο κεφάλαιο 4.2 των Λεπτομερών Οδηγιών CT-1 Δήλωση περάτωσης κλινικής δοκιμής_gr_10 Μαρτίου 2011 Σελίδα 25 από 25
© Copyright 2026 Paperzz