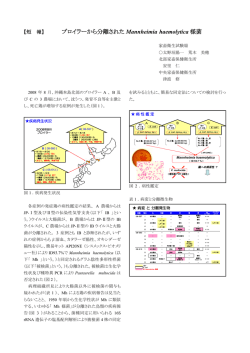
Application Note: Sequencing マルチプレックスの16Sメタゲノムを MiSeq® システムで高速解析 はじめに Greengenes4やRibosomal Database Project5など複数のデー タベースで入手可能です。分類においては、16S rRNA遺伝子 複雑な微生物群集のプロファイリング解析において、次世代 シーケンサ ーは 強力 なツ ール とな り つ つ あ り ま す。MiSeq システムは簡単操作のシーケンスワークフローを可能にし、 サンプルからデータまで迅速な解析が行えます。実績のある 1塩基合成反応( SBS: Sequence by Synthesis )テクノロジー を 採 用 し たMiSeqは、 次 世 代 シ ー ケ ン サ ー 上 位 機 種 で あ る HiSeq™ 2000システムと同等の高品質なデータを、ランあた り1Gb以上産出します。本アプリケーションノートでは、24 サンプルのPCR産物をインデックスを用いた150塩基のペア エンドでシーケンスを行い、末端部分を重ねたリードを使って 16Sメタゲノム解析を行いました。 の全長の代わりに、超可変領域のみをシーケンスすることで十 分な情報が得られます6,7。多くの微生物では、16S rRNA遺伝 子の4番目の超可変領域( V4 )は約254塩基配列からなり、配列 長の違いはわずか数塩基です。 V4領域増幅のながれ 16S rRNA遺伝子のV4領域をシーケンスするために、プライマー はV4領域周辺の保存性の高い領域をもとにデザインしました2。 MiSeqシステムでは150塩基のペアエンド解析ができ、ふたつ のリードの末端がオーバーラップすることで非常に高品質な 全V4領域の情報を得る事ができます( 図1 )。これらのプライ マーの末端にはMiSeqシステムで解析が可能なように、対応ア ダプターとインデックス用のバーコードが組み込まれていま す。この実験では、24種類の識別が可能なインデックス対応の プライマーを使い、24サンプルからV4領域を増幅しました。 MiSeqシステムのランには、これらの24サンプルを混合(プー ル)し、1回のランで同時に解析を行いました。 宿主関連と自由生活微生物群集間の既知の違いや、シーケンス データから得られる生物学的な結論は、どのシーケンスシステ ムでも高い再現性をもたらすはずです1, 2。シーケンス後の微生 物群集解析は、オープンソースのソフトウェアパッケージとし て複数の標準群集解析ツールを統合したQIIME( Quantitative Insights Into MicrobialEcology )パイプライン3を用いて行いま した。これらの結果は、これまでイルミナのGenome Analyzer™ システムで行われてきた高いスループットの微生物群集シーケ ンス解析が、MiSeqシステムでも同様に行え、これまで以上に 低コストで簡単なワークフローで実施できることを示しています。 MiSeqシステムでのシーケンスラン 異なるインデックスをつけた24サンプルは混合後、MiSeqシ ステム用の試薬カートリッジにアプライし、フローセルと一緒 にMiSeqシステムにセットしました。クラスター形成を行い、そ の後13サイクルのインデックス配列およびV4可変領域のシー ケンスを行いました。これらのステップはすべて自動化されて おり、約28時間で終了しました。 手法と結果 16S V4領域のシーケンス 16SリボゾームRNA( rRNA )遺伝子の配列多型は、微生物群集 データ解析 の分類学的多様性の特徴付けを行う際に広く使われています。 16S rRNA遺伝子の配列は9つの超可変領域と、その間に存在 する保存配列領域から構成されています。16S rRN遺伝子の 配列と超可変領域は、数多くの生物種で既に決定されており、 1次解析( 画像解析およびベースコール )はMiSeqシステム上 で行いました。クオリティフィルターした qseq ファイルは 、 QIIME8を用いてオフラインで解析しました。QIIMEはシーケン スの生データから論文投稿に必要な図を作成することができる 図1:V4領域増幅のながれとペアエンドリード P5 A. 515F V4 インデックス 806R P7 46 塩基の オーバーラップ SBSF (515F) 150 塩基 P5 インデックス V4 254 塩基 B. P7 SBSR (806R) 150 塩基 1600 1400 1200 1000 800 600 400 200 0 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160 170 180 190 200 210 220 230 240 250 A. 各サンプルのV4領域は515Fと806Rのプライマーを使い増幅しました。プライマーにはP5とP7の配列も組み込まれています。 46塩基のオーバーラップをも たらすペアエンドの150塩基のシーケンスランにより、V4領域の全254塩基情報を得ることができます。 B. V4領域の完全な254塩基配列をペアエンドリードで読み取った例。 (マトリックスとフェージングを修正後の)生強度を示しています。 Application Note: Sequencing 図2:QIIMEを用いた門レベルでの分類 100 90 発現の比率 (16S rRNA遺伝子配列の割合) 80 70 60 50 40 30 20 10 0 イヌ イヌの飼主(ヒト) Crenarchaeota Nitrospirae Euryarchaeota OP3 AD3 Planctomycetes Acidobacteria Proteobacteria Actinobacteria SC3 Armatimonadetes SC4 BRC1 SM2F11 Bacteroidetes SPAM CCM11b SR1 Chlamydiae Spirochaetes Chlorobi Synergistetes Chloroflexi TM6 Cyanobacteria TM7 Elusimicrobia Tenericutes Firmicutes Thermi Fusobacteria Verrucomicrobia GAL15 WPS-2 Gemmatimonadetes WS3 Lentisphaerae 土壌 微生物群集における16S配列領域をMiSeqとQIIMEを使って解析しました。イヌ、イヌの飼い主(ヒト)、土壌のサンプルから得られた結果は、門のレベルで分類し ています。 ように設計されており、リードのクオリティフィルターや、イン デックスサンプルの分類、操作的分類単位( OTU )の選定、分類 の割り当て、そしてアルファおよびベータの多様性解析も行う ことができます。図2は様々な環境から得られた24種類のサン プルの門レベルでの分類結果を示しています。これらのサンプ ルにはイヌ、イヌの飼い主(ヒト )、そして土壌サンプルを含み、 解析にはリード1( 5末端からのリード )を用いて行いました。 これらのサンプルはHiSeq 2000システムでも同様に解析され ましたが、どちらのシステムで得られた結果も高い再現性を示 すものでした(データは非表示)。 まとめ シンプルなインデックスの利用と大量データの産出が可能な MiSeqシステムは、微生物プロファイリングに適しており、また 迅速にサンプルから答えを導き出します。多様な微生物群集か らの16S rRNA遺伝子のV4領域をシーケンスした今回の結果で は、門レベルの分類ができました。QIIMEなどのオープンソー スのツールを使うことで、複雑な群集解析を行い、さらに論文投 稿に必要なデータも、サンプル入手後わずか数日でできるよう になりました。MiSeqシステムは今回の実験で示した例よりさ らに多くのサンプル、そして複数の16S rRNA可変領域を処理 できる能力をもっており、大規模なメタゲノム解析においても 十分なゲノム解析を可能にします。 参照 1. Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, et al. (2011) Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci USA 108:4516–4522. 2. Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, et al. (2011) Manuscript in preparation. 3. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman, FD, et al. (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7(5):335–336. 4. Desantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, et al. (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72(7): 5069–5072. 5. Cole JR, Wang Q, Cardenas E, Fish J, Chai B, et al. (2009) The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucl Acids Res 37:D141–D145. 6. Liu Z, Lozupone C, Hamady M, Bushman FD, Knight R. (2007) Short pyrosequencing reads suffice for accurate microbial community analysis. Nucl Acids Res 35:18. 7. Liu Z, DeSantis TZ, Andersen GL, Knight R. (2008) Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucl Acids Res 36:18. 8. http://qiime.org/tutorials/processing_illumina_data.html MiSeqシステムについての詳細は http://www.illuminakk.co.jp/product/system/miseq.shtml をご覧ください。 イルミナ株式会社 代理店 〒108-0014 東京都港区芝5-36-7 三田ベルジュビル 22階 Tel (03) 4578 - 2800 Fax (03)4578 -2810 www.illuminakk.co.jp 本製品の使用目的は研究に限定されます。 © 2014 Illumina, Inc. All rights reserved. Illumina, illumina Dx, BaseSpace, BeadArray, BeadXpress, cBot, CSPro, DASL, DesignStudio, Eco, GAIIx, Genetic Energy, Genome Analyzer, GenomeStudio, GoldenGate, HiScan, HiSeq, Infinium, iSelect, MiSeq, Nextera, NuPCR, SeqMonitor, Solexa, TruSeq, TruSight, VeraCode, the pumpkin orange color, the Genetic Energy streaming bases design は Illumina, Inc の商標または登録商標です。 その他の会社名や商品名は、各社の商標または登録商標です。予告なしに仕様を変更する場合があります。 Pub. No. 770-2011-J013 20MAR2014
© Copyright 2026 Paperzz