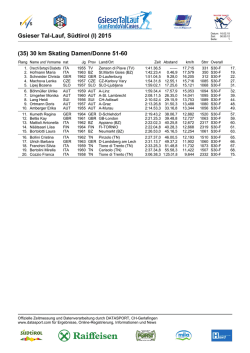
STUDI GENOTIPICI E FILOGENETICI DI ROTAVIRUS A (PoRV) E INFLUENZA A (SIV) VIRUS SUINI Zaccaria Guendalina CICLO XVII Tutor: Prof. Fabio Ostanello e Dott. Gabriele Vaccari Introduzione Rotavirus A (RVA) e Influenza A virus (IAV) sono agenti virali ad ampio spettro d’ospite, capaci di infettare molte specie animali compresi il suino e l’uomo. Sono virus a RNA a genoma segmentato che possono andare incontro a fenomeni di riassortimento tra la popolazione virale umana e quella animale con l’emergenza di ceppi nuovi emergenti capaci di diffondere nell’uomo. Il suino può avere un ruolo di serbatoio sia per quanto riguarda RVA che IAV. Lo studio ha l’obiettivo di evidenziare link molecolari, tramite analisi genomica e filogenetica, fra virus circolanti negli animali e quelli descritti in letteratura nell’uomo. E’ stato utilizzato il sequenziamento Sanger per il lavoro condotto sui Rotavirus, mentre per quanto riguarda lo studio su Influenza virus sono state settate le condizioni di sequenziamento dell’intero genoma virale mediante l’applicazione di tecniche di sequenziamento di nuova generazione (NGS) con piattaforma Ion Torrent PGM. Materiali e metodi Materiali e metodi L’RNA virale è stato estratto da campioni fecali suini risultati positivi per Rotavirus mediante ELISA. Ogni segmento del genoma è stato retrotrascritto ed amplificato con primers specifici mediante RT-PCR one step (QIAGEN OneStep RT-PCR Kit) ed i prodotti di amplificazione visualizzati mediante corsa elettroforetica su gel di agarosio. Le reazioni di sequenziamento sono state eseguite con il BigDye® Terminator v1.1 Cycle Sequencing Kit e le sequenze determinate con elettroforesi capillare su ABI 3130. Ad ognuno degli 11 segmenti del genoma di Rotavirus è stato assegnato un genotipo utilizzando RotaC version 1.0 (http://rotac.regatools.be.), un programma per la genotipizzazione di Rotavirus A disponibile su internet. L’RNA estratto è stato prima retrotrascritto poi amplificato mediante una multiplex PCR (PathAmpTM Flu A Reagents). Il genoma virale è stato sequenziato mediante piattaforma Ion Torrent PGM, un sequenziatore di nuova generazione. Il materiale amplificato è stato frammentato fino ad ottenere frammenti di circa 200 bp di lunghezza. Ai campioni analizzati sono stati legati dei “barcode” (1-16), degli oligonucleotidi con sequenza nota, che permettono il sequenziamento contemporaneo di più campioni in una sola corsa. La libreria così ottenuta è stata amplificata e sequenziata con lo Ion Torrent PGM. Allineamento e analisi del coverage sono state effettuate con il Torrent Suite Software. Tabella 2: campioni analizzati con Ion Torrent PGM. Dieci campioni sono stati corsi sia con il chip 314 sia con il 316. Risultati Risultati Abbiamo caratterizzato gli 11 segmenti dell’intero genoma di un ceppo (campione 2) isolato dal contenuto intestinale di un suinetto con diarrea (Tabella 1). L’analisi genotipica ha evidenziato che tutti i segmenti genomici fatta eccezione di NSP3 presentano un genotipo frequentemente riscontrato nella popolazione suina. Al contrario, il genotipo assegnato al gene della proteina NSP3 è un genotipo raro (T7), ad oggi assegnato a soli 7 isolati sia umani che animali. La sequenza di un singolo isolato H1N1pdm, ottenuta con il metodo NGS, è stata comparata a quella ottenuta con metodo Sanger: le sequenze mostrano un’identità nucleotidica del 100%. Abbiamo sequenziato con successo 36 isolati appartenenti a diversi sottotipi (H1N1, H1N2 e H3N2), collezionati in Nord Italia tra il 1998 e il 2012, correndo contemporaneamente su un unico chip fino a 16 campioni (Tab. 2). I dati sono stati analizzati in funzione di una sequenza di riferimento: per ogni campione l’analisi del coverage mostra una profondità di 20x per almeno il 99% della sequenza. In figura 2 è portato ad esempio il grafico di uno dei campioni analizzati che mostra la profondità del coverage della sequenza allineata ottenuta. Tabella 1: Genotipi assegnati agli 11 segmenti genomici del campione 2. RVA/Pig-wt/ITA/204BS/2010/G9P23 100 77 In ragione di questo risultato, abbiamo analizzato il segmento genomico che codifica la proteina NSP3 di altri 24 campioni, collezionati tra il 2009 e il 2010 da suinetti con diarrea in diversi allevamenti del Nord Italia. In totale 16 dei 25 isolati analizzati sono stati assegnati al genotipo T7 mentre solo 6 al genotipo T1, il genotipo della NSP3 più comunemente riscontrato nella popolazione suina. L’analisi filogenetica ha evidenziato che la maggior parte dei campioni con genotipo T7 clasterizza insieme e presenta una più stretta vicinanza ad un ceppo bovino, solo due campioni clasterizzano insieme ad un ceppo umano di probabile derivazione suina (Fig.1). RVA/Pig-wt/ITA/498BS/2010/GXP23 RVA/Pig-wt/ITA/622BS/2010/G9P6 NSP3 RVA/Pig-wt/ITA/492BS/2010/G5P23 RVA/Pig-wt/ITA/9BS/2009/G4P6 RVA/Pig-wt/ITA/5BS/2009/G5P13-P22 RVA/Pig-wt/ITA/10BS/2009/G9P23 100 RVA/Pig-wt/ITA/11BS/2009/G4P23 100 RVA/Pig-wt/ITA/12BS/2009/G4P23 RVA/Pig-wt/ITA/17BS/2009/G9P13-P22 T7 RVA/Pig-wt/ITA/519RE/2010/G5P23 RVA/Pig-wt/ITA/13BS/2009/G5PX RVA/Pig-wt/ITA/2CR/2009/G9P23 100 RVA/Pig-wt/ITA/16BS/2009/GxPx Bovine Bovine-like RVA/Cow-tc/GBR/UK/1973/G6P75 RVA/Human-xx/IND/mani-265/2007/G10P6 81 RVA/Hu/CHN/R479/2004/G4P6 Porcine-like RVA/Hu/BEL/BE2001/2009/G9P6 RVA/Pig-wt/ITA/4BS/2009/G5P6 99 RVA/Pig-wt/ITA/524BS/2010/G9P23 89 RVA/Pig-wt/ITA/6BS/2009/GXP6 RVA/Pig-wt/ITA/19BS/2009/G4P6 88 100 99 RVA/Pig-wt/ITA/8RE/2009/G4P6 RVA/Pig-wt/ITA/14BS/2009/G9P6 RVA/Pig-wt/ITA/3BS/2009/G9P6 98 99 RVA/Pig-wt/ITA/7RE/2009/G9P13-P22 RVA/Pig-wt/IND/RU172/2002/G12P7 100 92 100 RVA/Human-wt/BEL/B4633/2003/G12P8 T1 Figura 2: Coverage del campione 10169. Il grafico presenta in ascissa gli 8 segmenti genomici di Influenza A virus, mentre in ordinata il numero (log10) di letture di ogni singola base della sequenza allineata che è stata evidenziata durante il sequenziamento. Questo è un indice della validità e veridicità della chiamata della base. I segmenti più corti hanno un coverage maggiore rispetto a quelli lunghi. RVA/Human-wt/BEL/B3458/2003/G9P8 RVA/Human-tc/USA/Wa/1974/G1P1A8 RVA/Human-xx/IND/mcs-13/2007/G9P6 RVA/Pig-tc/MEX/YM/1983/G11P97 RVA/Pig-tc/USA/Gottifried/1983/G4P6 RVA/Pig-tc/USA/OSU/1977/ G5P97 100 RVA/Pig-tc/VEN/A253/1988/G11P97 100 100 RVA/Pig-tc/VEN/A131/1988/G3P97 RVA/Human-tc/USA/DS-1/1976/G2P1B4 100 T2 RVA/Human-wt/RSA/GR10924/1999/G9P6 RVA/Pigeon-tc/JPN/PO-13/1983/G18P17 T4 PB2 PB1 PA HA NP NA M NS 0.1 Figura 1: albero filogenetico del gene della proteina NSP3. I campioni analizzati sono evidenziati nella cornice rettangolare, in rosso quelli appartenenti al genotipo T7 e in verde quelli al genotipo T1. I ceppi sono stati contrassegnati in base alla specie da cui sono stati isolati: suino uomo bovino Conclusioni Il sequenziamento dell’intero genoma è un importante strumento per comprendere l’evoluzione e l’ecologia di agenti virali molto variabili come quelli presi in esame. Mettendo a confronto i risultati ottenuti nei due lavori nel corso degli ultimi 6 mesi, sono evidenti i vantaggi dell’utilizzo delle tecniche di NGS. Lo sviluppo di protocolli con metodo NGS, che permettano il rapido sequenziamento di un gran numero di isolati virali, potrebbe dimostrarsi fondamentale in situazioni critiche di epidemie. Tale metodologia necessita tuttavia di una validazione tramite confronto con il sequenziamento Sanger che rimane il metodo di riferimento. Presentazioni orali: •“Full-length genomic analysis of porcine Rotavirus strains isolated from pigs with diarrhoea in Northern Italy” presentato all’11th National Congress of the Italian Society for Virology ad Orvieto. Poster: •“Next generation sequencing to study the molecular evolution and transmission dynamics of Influenza A virus in pigs” presentato al congresso Arbo-zoonet “Joint Conference on Emerging and Re-emerging Epidemics Affecting Global Health” ad Orvieto.
© Copyright 2026 Paperzz