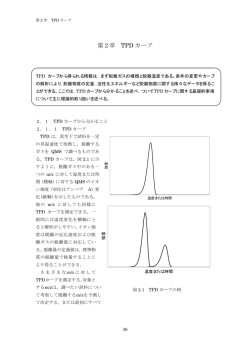
第1章 昇温脱離法および関連することがら 第1章 昇温脱離法および関連することがら 昇温脱離法(TPD)は試料を高真空下で加熱して発生する気体を分析して,試料に吸着している 物質や試料の表面近傍の状態を解析する方法である。ここでは,TPD およびそれに関連する事 項として,まず真空,質量分析法,表面吸着や熱分解反応などについて述べる。ついで,TPD の 原理,装置の概略および TPD カーブについて述べる。 1.1 はじめに 物質の熱による変化を調べる方法として,熱分析法はひろく活用されている。その中で も加熱により試料から発生したガスを分析する方法を,発生気体分析(Evolved Gas Analysis:EGA)と総称している。この方法は加熱条件や分析手法により様々なものがあ る 。 試 料 の 加 熱 は 高 真 空 下 や 大 気 圧 下 で 行 い , 発 生 し た ガ ス は 質 量 分 析 法 ( Mass Spectrometry : MS ) や フ ー リ エ 変 換 赤 外 分 光 法 ( Fourier Transform Infrared Spectroscopy:FT-IR)などにより分析するものが多い。その中で,制御された条件下で試 料 を 加 熱 し , 試 料 か ら 脱 離 す る ガ ス を 分 析 す る 方 法 を , 昇 温 脱 離 法 ( Temperature Programmed Desorption:TPD)という1-4)。加熱温度を制御して行うのでこの名称がつけ られた。または,TDS(Thermal Desorption Spectroscopy)ともいう。両方の名称がこれ まで使用されてきたが,最近では前者が一般的に用いられるようになってきている*1。 1.2 昇温脱離法 1.2.1 始まりと発展 TPDは,もともと試料の吸着ガスを分析する方法として発展し,すでに 1963 年に触媒の 分析に用いられて,触媒の表面解析では有用な手法となっている。さらに半導体材料の吸 着物質の解析10-13)にも応用され,表面の清浄度の解析に用いられている。さらに現在では, 吸着物質の解析のみならず,物質の表面や表面近傍の状態,薄膜の吸着物質や表面および 内部の状態を解析する方法として広く用いられ,固体材料の表面状態およびその近傍を解 析する方法として多用されている14-17)。たとえば,単結晶表面への吸着物質の解析14),ゼオ ライトや金属酸化物の固体酸塩基性の解析15,16)や土壌改良や揮発性有機化合物(Volatile Organic Compounds:VOC)*2)の解析17)などに用いられている。 1 昇温脱離法の文献では,TPDとTDSの使用頻度は約 4:1 である。 2大気中に排出され,又は飛散した時に気体である有機化合物であり,浮遊粒子状物質やオキシダントの生 成要因となるもの。 1 第1章 昇温脱離法および関連することがら 1.2.2 昇温脱離法(TPD) さて,TPD においては,制御された方法で加熱するのであるが,昇温速度を一定とする ものが多い。その雰囲気はキャリアガスを流した常圧または高真空下であるが,後者がよ り多い。放出されるガスの分析は,初期では,ガスをガスクロマトグラフィーや質量分析 計により分析してきたが,最近では,四重極質量分析計(Quadrupole Mass Spectrometer: QMS)を一般には用いる。だから,正確には TPD-MS である。 従って,試料表面の吸着ガス,試料の表面やその近傍からガスとして放出される成分を 高精度で分析でき,10−10モルという微量分析が可能である。触媒や半導体の解析ではその 微量分析の特徴を活かしたものである。TPDの解説は文献1-6,18)もあわせて参照されたい。 なお,加熱を大気圧下で行い,熱重量分析(Thremogravimetry:TG)と同時に発生す るガスを分析する方法もある20)。分析方法として質量分析法(MS)を用いるものをTG-MS といい,同様にフーリエ変換赤外分光法(FT-IR)を用いるものをTG-FTIRという。TPD と類似の分析法である。 1.2.3 セラミックスへの応用 一方,セラミックスおよびその薄膜は,多くの機能性を発現し,機能性材料として有用 である。その表面および近傍の状態や吸着種の解析は,セラミックスの材料化学として重 要である。セラミックスは焼成や焼結プロセスを経てバルク体や成形体となる。また,セ ラミックスの化学的作製法であるゾルーゲル法においては,一般に焼成プロセスを経てセ ラミックス粉体や薄膜を作製する。従って,TPDをセラミックス研究に用いることにより, 焼成プロセスの解析や表面およびその近傍の状態などについての知見が得られる18,19) 。た とえば,窒化ケイ素やAlNの製法による表面状態の変化に関する研究例がある18,19)。 筆者らは,TPDを用いて,ゾルーゲル法によるセラミックス薄膜の焼成過程の解析や吸 着種の解析を行い,その有用性を認識している21-23)。しかし,他分野における研究に比べる とセラミックスに関するTPDの応用は活発とは言えないのが現状である。 TPD(TPD-MS)は,上で述べたように高真空下で試料を加熱し,脱離したガスを QMS で測定するものである。そこで,以下では,まず,TPD に関連する事項として真空や質量 分析法について述べる。TPD ではこれらが用いられているからである,ついで,反応速度 論,表面吸着・脱離や熱分析の一般的知見を概観する。TPD の基礎であるからだ。最後に, 昇温脱離装置について説明し,TPD カーブについて述べる。 2 第1章 昇温脱離法および関連することがら 1.3 真空 1.3.1 真空 われわれは大気圧下で生活している。われわれの体は空気からの圧力(大気圧)を受け ているが,通常はその力を感じることはない。大気は酸素や窒素などの分子が充満してお り,それが運動し物体に衝突することにより大気圧を生み出している。空気を容器に密封 してそこから空気を除去すると圧力が減少して真空が得られる。真空は,空気を形成する 分子がきわめて少ない状態である。TPD は真空下で試料を加熱する分析装置であるから, 以下では真空について概略を述べる。 1)単位 圧力の単位についてまず述べる。SI単位では圧力はパスカル(Pa N/m2)を用いる。圧 力は単位面積あたりに作用する力としてあわらされ,1Paは 1m2あたりに 1Nの力が作用し た圧力である。従来との単位との関連および大気圧との関係を表 1.1 に示す。 表 1.1 Pa 単位と従来の単位との関係および大気圧の Pa 単位による大きさ 事項 1Pa 1N/m2 標準大気圧(1 気圧) 1.01325×105Pa 標準大気圧(1 気圧) 1013.25hPa(ヘクトパスカル) 1torr(トール) 133.322Pa 1bar(バール) 105Pa h=102 1 モルの気体は,0℃,1 気圧で 22.4Lの体積を占め,6.023×1023個(アボガドロ数)の 分子が含まれる。この中では分子が無秩序に運動しており,分子は衝突しあっている。気 体分子が衝突しないで飛べる距離(つまりある衝突から次の衝突までの距離)を平均自由 工程といい大気圧下では 10−5cm程度である。 2)真空の種類 容器内に気体を閉じこめ,気体を取り去っていくと真空になり圧力が低下する。その程 度により真空は区分されている。表 1.2 に真空の種類とその気体分子密度および平均自由工 程を示す。 3 第1章 昇温脱離法および関連することがら 表 1.2 真空の種類とその気体分子密度および平均自由工程 気体分子密度 平均自由工程 (個/cm3) (cm) 大気圧(105)∼102 3×1019∼3×1016 3×1016∼3×1013 高真空 102∼10―1 10−1∼10―5 1×10−5∼1×10−2 1×10−2∼10 3×1013∼3×109 10∼1×105 超高真空 10−5∼10―8 3×109∼3×106 1×105∼1×107 極高真空 <10―8 <3×106 >1×107 真空の種類 圧力(Pa) 低真空 中真空 1.3.2 λ 真空をつくる 真空をつくるには,容器内の空気を除去するのだが,これには真空ポンプを用いる。 低真空をつくるには一般的には油回転ポンプ(ロータリーポンプ Rotary Pump:RP) を用いる。RP にはいくつかの種類があるが,いずれも作動部が油槽に浸されている。その ため,真空にしたい容器内に油蒸気が残ってしまう。そのため,汚染のない高真空をつく るにはさらにもう一段真空度を高めるポンプと併用して用いられる。RP の例を図 1.1 に示 す。これは回転翼型であり,ロータが回転することにより吸気口から空気を取り入れる。 ロータがさらに回転して排気口から空気を除去する。この繰り返しにより真空をつくる。 高真空をつくるポンプにはいくつかあるが,最近ではターボ分子ポンプ (Turbo-Molecular Pump:TMP)が用いられる。TMPには,モーターにより高速回転す る動翼(ファン)と,本体に固定翼が取り付けられている。1 分間あたり数万回転で高速回 転する動翼で吸気口から取り入れた気体分子を弾き,それを固定翼にあてて次の動翼へ送 る。これを繰り返して,最終的に排気口へと気体を押し出す。TMPの特徴は,排気速度が 気体の種類によらず,油を使用せず,清浄であり,10−10 Pa程度という高い真空度が得られ ることである。 図 1.1 油回転ポンプ(回転翼型)24) 4 第1章 昇温脱離法および関連することがら 図 1.2 ターボ分子ポンプ24) 5 第1章 1.4 昇温脱離法および関連することがら 質量分析(四重極質量分析計) 1.4.1 質量分析 TPD では,脱離物質の同定には一般的には四重極質量分析計(Quadrupole Mass Spectrometer:QMS)が用いられる。その原理である質量分析法(Mass Spectrometry: MS)は次のプロセスで物質を検出する。すなわち, ① 調べたい試料を高真空下でガス化する, ② ガスに一定の電子流をあてると試料中から電子がたたきだされて,カチオンラジカル が生成し,さらにそれが開裂した断片(フラグメントイオン)が形成される(電子衝 撃イオン化法), ③ これらの生成物はその質量(m)と電荷(z または e)の比,すなわち質量電荷比(m/z) として分離され,それぞれの m/z ごとにイオン電流値として検出される。 1.4.2 四重極質量分析計(QMS) 四重極質量分析計(QMS)では,③の分離プロセスに,直流と高周波交流をあわせた電 圧を印加した四重極質量分離装置を用いる。その概念図を図 1.3 に示す。 イオン化 + − − + イオンの進行 図 1.3 四重極質量分離装置の概念図 QMS 内には約 20cm の 4 本のロッドが設置され,隣り合うロッドには異なる電位が印加 され,対抗するロッドには同一の電位が印加されている。ここで印加される電位は高周波 電位に直流電位を重ねあわせたものである。ガスが左下から導入され,電子線衝撃により イオン化される。イオンが四重極質量分離装置内(4 本のロッドの空間内)に入ると,印加 される高周波により振動する(図 1.3 の波線はそれを模式的に示す)。このとき,ある電位 においては特定のイオンのみが安定に振動してロッド内を通り抜ける。従って,ある電位 では,特定の m/z のイオンが検出管である電子倍増管に入っていく。電位を変化すること により,異なるイオンが電子倍増管に入る。このようにしてイオンが検出される。 6 第1章 昇温脱離法および関連することがら 1.5 反応速度論 表面吸着物質の脱離反応や薄膜やバルク体の熱分解反応は,反応速度論的に解析するこ とが多い。そこで,ここではその基礎となる化学反応速度論について簡単に述べる。 1.5.1 反応速度の解析 1)反応速度 以下の反応を考える。 A → B + C (1) これは A が B と C に変化するという反応である。反応を考える尺度の一つに反応速度が ある。すなわち,A の減少する速さ,または B,C の増加する速さである。数式で表すと以 下のとおりとなる。 d [ A] dt d [B ] vb = dt d [C ] vc = dt va = − (2) (3) (4) Aの濃度が少しの時間(dt)で少し減少する(−d[A])のだから,これを単位時間で割 ると単位時間における濃度の変化となる。これは反応速度である。すなわち,上式となる。 ここで,va=vb=vc=vである(vはこの反応の反応速度)から,(5)式が得られる。 v=− d [A] d [B ] d [C ] = = dt dt dt (5) ところで,反応を一般化すると下式になる。 A → nB + C (6) この反応では,A が 1 個から n 個の B と 1 個の C が生成するのだから − d [A] 1 d [B ] = dt n dt (7) 7 第1章 昇温脱離法および関連することがら となる。従って,このときの反応速度 v は,(8)式のとおりである。 d [A] 1 d [B ] d [C ] = = dt n dt dt v=− (8) だから,反応速度式は一般的に(9)式となる。 v= 1 d [X ] n dt (9) ここで,X は生成する物質であり, [X]はその濃度を表し,n は物質 X の量論係数である。 2)反応次数 上の反応では,反応速度 v を A の濃度([A] )との関係で考察したが,いつもこうとは限 らない。A の濃度の 2 乗や 3 乗に比例してもよい。一般的には反応は A の濃度の n 乗に比 例すると考えられる。だから,反応速度 v は(10)式で記してよい。 v = k [ A] n (10) ここで,k を速度定数といい,n を反応次数という。n は整数でなくてもよい。上の反応 は,「[A]について n 次である」という。 3)反応速度の積分形 反応速度は上では 2 つの式(9,10 式)で表されている。 v=− d [ A] dt (9) v = k [ A] n (10) (10)式で n=1 として以下の議論を進める。 この 2 つの式より(11)式が得られる。 − d [ A] = k [ A] dt (11) 8 第1章 昇温脱離法および関連することがら (11)式は 1 d [A] = − kdt [ A] (12) と置き換えられる。この式は積分できる。反応の開始時(t=0)のAの濃度を「A」0とし, 反応が時間t進んだときのAの濃度を[A]とする。上式を時間t=0 からtまで積分すると,(13) 式になる。 [ A] ∫[ ] [A]d [A] = − ∫ kdt A 1 (13) t 0 0 (13)式は,反応速度式の積分形である。 従って,(14)式が得られる。 ln [ A] [A]0 = − kt (14) これは [A] [A]0 = e − kt (15) であるから, [A] =[A]0 e − kt (16) となる。 (16)式は反応時間tにおけるAの濃度([A])は初期濃度[A]0にe−ktをかけたもの となる。e−ktは 1 より小さいから,[A]は 1 よりも小さくなり,時間tに対して指数関数的 に減少する。 9 第1章 昇温脱離法および関連することがら 1.5.2 活性化エネルギー 1)活性化エネルギー(アレニウスの式) 以下の反応((17)式)をもう一度考える。 A → B + C (17) 反応物である A も生成物である B と C も安定体であると,一般的には考えられる。しか し,その安定な A が変化するのだから,一般的には反応は外の系からエネルギーが投入さ れて起こるのである。たとえば,加熱や光照射などである。そうすると,A にエネルギーが 投入されてある活性状態となって変化すると考えられる。 アレニウス(Arrhenius)*3は,実際の化学反応の進行状況や理論的考察に基づき,活性 状態(活性複合体)について考察した。上の反応が分子の反応であるとする。活性状態は反 応物のAよりエネルギーが高い状態であると仮定してよい。また,自然界における粒子の分 布は,ボルツマン分布に従う。反応全体の分子数(X)に対してある状態の分子数(Y)は, その状態のエネルギーをEaとすると(18)式となる。 E − a Yの数 = e RT Xの数 (18) ここで,R は気体定数,T はそのときの絶対温度である。だから,(19)式になる。 E − a 活性複合体分子の数 = e RT 反応物分子の数 (19) ここで,Eaは活性複合体のエネルギーであり,これを活性化エネルギーとする。アレニウ スによれば反応速度は,活性複合体分子の数に比例する。だから,反応速度(v)は(20)式 であらわされる。 v = α × (活性複合体分子の数 ) = α × (反応物分子の数 ) × e − Ea RT (20) ここで,αは比例定数である。従って,(21)式が得られる。 Svante August Arrhenius (1859∼1927) スウエーデンの物理化学者。電解質の解離(電解質は溶液中 では陽イオンと陰イオンに解離する)をはじめて提唱したが,長く学会には受け入れられなかった。アレ ニウスの式を提出し,化学反応速度論の発展に貢献した。 3 10 第1章 昇温脱離法および関連することがら v = A× e − Ea RT × (反応物の濃度 ) (21) (21)式から,次の関係式(22)が得られる(反応次数の項 k = A× e − (10)式参照)。 (22) Ea RT ここでkは速度定数,Rは気体定数,Tは絶対温度である。この式をアレニウスの式といい, Aを頻度因子,Eaを活性化エネルギーという。この 2 つを反応のアレニウスパラメーターと いう。Eaは模式的には図 1.4 のように理解される。 エネルギー 活性化状態 Ea A B C 反応の進行 図 1.4 反応の進行と活性化エネルギー また,頻度因子は,活性化状態に関係しており,反応分子どうしの衝突のしやすさや活性 化分子の状態をあらわしている。 2)活性化エネルギー(アレニウスプロット Arrhenius plot) 上でも述べたが,アレニウスの式は(22)式のとおりである。 k = A× e − Ea RT (22) これを書き直すと(23)式が得られる。 11 第1章 昇温脱離法および関連することがら ln k = ln A − Ea E 1 → ln k = ln A − a × RT R T (23) だから,1/Tに対してln kをプロットする。これをアレニウスプロットという*4。その傾き は−Ea/Rとなる。従って,アレニウスプロットから活性化エネルギーを求めることができ る。 アレニウスプロットから活性化エネルギーを求めるときに,注意しなければならない以下 のことがらがある。 ① アレニウスプロットの前提となっているのは,反応が単一の反応ということである。 ② アレニウスプロットを行うと,それは直線にならねばならない。プロットの一部が直線 であるが,他は曲がっていたり,段差になっていることもある。そのような部分は異な った反応が起こっていると判断しなければならない。そのような部分をEaの算出に用い ることはできない。 ③ Eaの算出にあたってはできるだけ多くの温度領域を測定して直線になる部分を用いる べきである。また,求めたEaについては,温度領域や濃度・反応率をできるだけ明確に しておくべきである。 3)活性化エネルギー(Ea)の解釈 Eaは上で述べたように活性複合体と反応物とのエネルギーの差異であると考えてよい。 Eaはいまの反応では,AがBとCに変化するときに経由するある活性な状態のエネルギーに 対応している。だから,ある活性状態を経由して反応が進行することは理解できる。 前提条件として確認しておきたいのは,ここで調べている反応は,単一の素過程で進行し ているということである。 しかし,反応の進行経路は単純ではない。A が B と C になる反応は,反応条件や環境に よって変わることが予想される。つまり,図 1.5 のようになると考えられる。 だから,同じAがBとCになる反応でも,反応条件や環境によって経路やEaが変化すると 考えられる。 4 アレニウスプロットは,1/Tに対するln kのプロットのことである。つまり,温度(絶対温度)の逆数に 対して反応速度(正確にはln k)をプロットするもののことである。 12 第1章 昇温脱離法および関連することがら 従って,Eaを求めるときは, ① 反応条件や環境を決めておく。 ② それらを比較・吟味するときには条件や環境が同じものどうしを比較する。 ③ それに加えて,その反応を調べている測定機器の問題もあるので,注意する。 ④ つまり,反応が同一でも機器により感度や測定量が異なるので,得られるデータが異な ることも充分に考えられるのである。 とはいえ,反応を考察するときには,活性化エネルギー(Ea)や頻度因子(A)は重要な ものであり,それらを用いることにより反応が詳細に考察できるのである。 エネルギー 活性化状態 A B 反応の進行 図 1.5 活性化状態のいろいろ 13 C 第1章 昇温脱離法および関連することがら 1.6 固体表面への吸着および脱離 1.6.1 固体表面とは 固体表面は,さまざまな物質が吸着,脱離や化学反応が行われる場である。表面は,固 体と気体(固相と気相)との界面である。表面には固体を形成する原子・分子・イオンが 存在しており,理想的にはそれらが一つの平坦面を形成している。結晶であれば規則性を もって配列している。しかし,このような表面を作製することは一般的にはたいへん難し い*5。現実に得られる結晶表面の例を図 1.6 に示す。表面は,テラス,ステップ,キンクや さまざまな欠陥からなる。テラスは原子レベルで平坦な面であり,ステップは段差,キン クはステップの曲がり角であり反応性が高い。欠陥は点欠陥などである。このような表面 で,原子・分子・イオンなどの吸着・脱離・化学反応が行われる。 図 1.6 結晶表面の構造25) 表面は界面であるから厚みはない。しかし,実際に表面に関することがらを研究する ときには,表面はある厚みを持つことになる。表面への吸着・脱離,触媒反応,センサー, や表面張力を取り扱うときは,最表面の 1∼数原子(分子やイオン)程度の厚さを考慮すれ ばよい。薄膜では,単分子∼数μm 程度の厚さが対象となる。表面近傍ということになる と,数μm∼10μm 程度であろう。それより厚いと表面とは言い難いだろう。 5 雲母(マイカ)は層状構造を持つ結晶である。マイカの層の表面はこのような理想表面に近い構造であ る。 14 第1章 昇温脱離法および関連することがら 1.6.2 表面吸着 1)表面構造と吸着 固体表面に物質が接近して吸着することを考察する。このときは,ポテンシャルエネル ギー図(図 1.6)を描いて考察する。 図 1.6 固体表面への吸着に関するポテンシャルエネルギー 吸着物質が固体から充分に離れていればポテンシャルエネルギーはゼロである。吸着物 質が表面に接近するとエネルギーは減少する(安定化する)。ある距離(r0)でその値は最 小値(ΔEa)となり最も安定化する。この状態が吸着である。それよりさらに接近すると 斥力が働きエネルギーは上昇する。ΔEaは吸着のエンタルピーであり,脱離の活性化エネ ルギーでもある。 2)物理吸着と化学吸着 ところで,吸着には物理吸着と化学吸着がある。物理吸着は,吸着物質がファンデアワ ールス力のような弱い相互作用で固体表面に吸着するものである。化学吸着は,吸着物質 と固体間に化学結合が形成されるものであり,結合力は大きい。物理吸着のエンタルピー は約−25KJ/mol 以下(絶対値の値として小さい)であり,後者のそれは約−40KJ/mol 以 上(絶対値の値として大きい)といわれている。これは一概にはいえない場合もあるが, 一応の目安としてよい。 吸着物質がどの程度固体表面に吸着しているかは,重要な情報であり,表面被覆率また は表面濃度といい,θであらわす。 吸着に関しては,ラングミュア(Langmuir)やベット(BET)の吸着理論があるが割愛 する。 15 第1章 昇温脱離法および関連することがら 1.6.3 物理吸着 物理吸着は,吸着物質がファンデアワールス力などの弱い相互作用で吸着しているもの である。吸着力は物質と固体との相互作用によるが,吸着分子は固体上に複数の層を形成 している。固体により近いものほど吸着力は強いが,離れるほど弱くなる。これは TPD で 解析可能である。図 1.13 に物理吸着水の TPD カーブの例を示す。 1.6.4 化学吸着 化学吸着は,吸着物質と固体との化学結合の形成によるものである。固体表面との結合 によるのだから,化学吸着層は 1 層である。化学吸着には,吸着分子が解離しないで吸着 するものと解離して吸着するものがある。それを図 1.7 に示す。 図 1.7 化学吸着のポテンシャルエネルギー a:物理吸着,b:解離しない化学吸着, c:解離を伴う化学吸着26) 吸着分子が固体に接近するとまず物理吸着となり(図 1.7 a),ΔEp分だけ安定化する。 解離しない化学吸着の場合(b)では,さらに分子が固体に接近して化学結合しより安定に なる。脱離の活性化エネルギーは吸着のエンタルピーに等しく(ΔEa),ポテンシャルゼロ から最小ポテンシャルまでのエネルギーである。これは物理吸着の場合より大きい。 それに対して,解離を伴う化学吸着(c)では(ここでは等核分子を考える),最初の物理 16 第1章 昇温脱離法および関連することがら 吸着は上と同様であるが,化学吸着のとき解離するので,解離エネルギーも考慮する。1 分 子の解離エネルギーを△EDとすると,1 原子の解離エネルギーは△ED/2である。1 分子 の脱離の活性化エネルギーを(△E0)mとすると,1 原子の脱離の活性化エネルギーは, (△E0)mに△ED/2を加えた値となる。この場合,脱離の活性化エネルギーは 1 分子の脱 離の活性化エネルギー(△E0)mとして考察してよい。 化学吸着ではもう一つ考慮しなければならない吸着がある。それを図 1.8 に示す。この場 合は,物理吸着から化学吸着へ移行するときに活性化状態を経由する。図ではΔEa*を経由 してΔEa分だけ安定化する。この吸着は反応温度に対して複雑な依存性を示す。このとき の脱離の活性化エネルギーはΔEaにΔEa*を加えた値となる。 図 1.8 活性化状態を経由する化学吸着 化学吸着に関しても TPD で解析可能である。図 1.14 に化学吸着水の TPD カーブの例を 示す。 17 第1章 昇温脱離法および関連することがら 1.6.5 表面吸着物質の脱離に関する反応速度論 1)ポラニーウィグナー(Polanyi-Wigner)式 固体表面に吸着している物質の脱離に関して,化学反応の速度論を適用して考察できる。 吸着している物質の量を表面濃度(または被覆率) (θ)としてあらわす。その脱離速度(ま たは脱離率)は表面濃度の時間あたりの脱離量と考えてよい。つまり,表面濃度θを時間 t に対して微分したものとしてよい。すなわち,脱離速度を v(t)とすると(24)式となる。 v (t ) = − dθ dt (24) (24)式を用いると,化学反応速度式((10)式)は(25)式となる。 v (t ) = − (25) dθ = k nθ n dt ここで,nは反応次数,knは速度定数であり反応次数の寄与を含む。(10)式では濃度は[A] として表記してあるが表面濃度θになり,速度定数kはknとなるので,(10)式は(25)式にな る。 速度定数は(22)式で示されていたが,それをこの脱離反応にあわせて記すと,(26)式にな る。 kn = ν ne − Ea RT (26) ここで,υnは頻度因子((22)式ではA),Eaは脱離の活性化エネルギー,Tは絶対温度であ る。(25)式と(26)式から,(27)式が得られる。 v (t ) = − − a dθ = ν nθ n e RT dt (27) E (27)式は,ポラニーウィグナー(Polanyi-Wigner)式といい,表面物質の脱離に関する基 本式である。記号について改めて寄与も含めて記す。υnは頻度因子であり温度と表面濃度 の寄与が含まれる。Eaは脱離の活性化エネルギーであり表面濃度の寄与が含まれる。nは反 応次数であり表面濃度の寄与が含まれる。θは表面濃度(表面被覆率),Rは気体定数 (8.31451J/Kmol)*6,Tは絶対温度である。 6 2002 年のCODATA(Committee on Data for Science and Technology)勧告によれば,Rは 8.314472J/mol 18 第1章 昇温脱離法および関連することがら 2)0 次,1 次,2 次の脱離反応 n=0 のときは 0 次反応であり脱離速度は表面濃度に依存しない。吸着層が充分に厚いと きに観測される。n=1 のときは 1 次の脱離反応で,表面濃度に比例して脱離速度は変化す る。また,n=2 のときは 2 次の反応であり,脱離速度は表面濃度の 2 乗に比例する。それ らの例を図 1.9 に示す。固体表面に物理吸着したH2Oの脱離は 1 次反応である。それに対し て化学吸着した水は固体表面にOHとして結合している。隣り合う 2 つのOHが反応して H2Oが脱離する反応が 2 次の脱離反応である。 H2O(気体) 脱離 H2O H2O H2O H2O H2O H2O H2O H2O H2O 固体 0次反応 脱離 H2O(気体) 物理吸着 H2O 固体 1次反応 H2O(気体) 脱離 化学吸着 OH OH O 固体 固体 2次反応 図1.9 固体表面からのH2Oの脱離反応 と修正されている。少数点 3 ケタ目まではこれまでと同じである。 19 第1章 1.7 昇温脱離法および関連することがら 熱分析 1.7.1 固体の熱分解反応 1)熱分解反応 固体に熱を加えると,分解,分解を伴う溶融,溶融,昇華および焼結反応が起こる。焼 結は結晶のごく一部が溶融状態を 経て焼き締まるプロセスである。 分解や分解を伴う溶融の結果,生 成物(結晶を含む)が得られる。 ここでは,固体の熱分解反応に ついて,その反応速度論による取 扱いについて述べる。 固体の熱分解反応は,固体に熱 を加えて起こる変化(分解)であ る。その反応は,固体の熱による 図 1.10 一定温度下における熱分解反応の進行27) 分解率*7 (α)を,一定温度下に おいて時間を変化したり,または温度を変化(一般的には一定の昇温速度で温度を変化さ せる)して調べる。図 1.10 に一定の温度(定温)条件下におけるαを示す。反応は,反応 の開始(A)から誘導期(B)を経て,Cで加速される(加速期)。Bでは反応核が生成して, Cで核が成長して反応が進行する。Dは反応速度が最大のポイントであり,この後減速し(E 減速期),Fで反応が終了する。 熱分解反応は,固体中にまず反応核が生成することによりスタートする。反応核は,微 小な活性領域であり,反応物中で微小領域が生成物に化学変化または組成変化したもので あるか,反応物中の生成物の微小な結晶核である。核の成長は,核と反応物との界面で化 学反応が進行することによる。そうすると,反応物と生成物の境界に反応界面ができ,そ れが進行することにより反応が進行する。 2)反応機構 反応界面では,反応物内の結合の切断,生成物の核発生と成長など,複雑な反応が,同 時にまたは逐次的に進行する。だから,これらを総合的にまとめた考察が必要となる。反 応界面で起こる反応は,いくつかの反応機構モデルが提案されている30)。反応がn次反応で あるフォーマル(Formal)型(Fn),界面における反応が律速である界面律速(Phase boundary reaction)型(Rn),化学種の拡散が律速である拡散律速(Diffusion)型(D1 ∼D5),核形成―成長(Nucleation and nuclei’s growth)型(An),自動触媒(Autocatalysis) 型(B1)であり,それぞれに速度論的モデル関数がある。それを表 1.3 に示す。 7 反応率ともいう,以下では「反応率」を用いることが多い。 20 第1章 昇温脱離法および関連することがら 3)化学反応速度論と固体の熱分解反応 ここで,化学反応速度論と固体の熱分解反応の違いについて記しておく。化学反応速度 論は,特に断っていないが,反応は溶液系で,均一系であり,反応は単一の素過程で進行 している。それに対して,固体の熱分解反応では,対象となる物質は固体であり,反応が 局所的に起こる不均一系であるところが異なる。このことにより,固体の熱分解の特徴が みられる。 1.7.2 固体の熱分解における反応速度 固体の熱分解は不均一反応であり,その場合には,反応速度 v は反応率αの時間微分 (dα/dt)としてあらわされる。上で述べたように固体の熱分解はいくつかの反応が起こっ ており,律速反応も様々である。そこで,いろいろな条件において反応速度が調べられた。 それぞれの律速反応に対応して反応速度式が検討された。それらを総合して,熱分解の一般 的な反応速度式が示されている。Sesták-Berggren式(28,29 式)である28,29)。 dα m n p = k (α ) (1 − α ) [− ln (1 − α )] dt dα m n または = k (α ) (1 − α ) dt (28) (29) ここで,k は速度定数である。下の式は,上式で p=0 とした場合と同じである。 ところで,上式で m=p=0 とおくと dα n = k (1 − α ) dt (30) となる。これは n 次の反応速度式であり,反応物が n 次で単純に熱分解する式となる。上 式を様々に変形することにより,いくつかの反応速度式を示すことができる。 上式の右辺は複雑であるが,αを変数とする関数 f と考えることができる。関数 f を熱分 解反応における速度論的モデル関数という。上で述べたいくつかの反応機構に対するモデル 関数を表 1.3 に示す。 21 第1章 昇温脱離法および関連することがら 表 1.3 固体の熱分解における反応機構およびモデル関数30) だから,(28)式や(29)式は,(31)式とすることができる。 dα = kf (α ) dt (31) これは微分型であるが,積分型で示すと,速度論的モデル関数として g(a)を積分型で定義 して g (a ) = ∫ α 0 dα = kt f (α ) (32) で示される。固体の熱分解においてもアレニウスの式((22)式))は成立する,と考えてよ い。 k = A× e − Ea RT (22) だから,(31)式は,(33)式となる。 − a dα = A × e RT f (α ) dt E (33) 22 第1章 昇温脱離法および関連することがら (33)式は,固体の熱分解が単一の反応で進行する場合に,基本となる反応速度式であり,理 想的にはすべての熱分解反応に適応可能である。 1.7.3 反応速度の解析 固体の熱分解における反応速度の解析は, ①熱分解の反応速度式を様々に変形して実験データを解析しやすくする, ②実験条件を変えてデータを取得する, ③変形した式に代入する,または式から導入される方法でデータをプロットする, ④EaやAを求める, である。 熱分析においては,いくつかの反応速度の解析法が提案されている。解析法は大きく分 けて,等温的方法と非等温的方法がある。それらはさらにいくつかの方法に分かれる。 このような方法によって活性化エネルギー(Ea)が求められるが,Eaは,測定条件,測 定機器や解析方法により異なる値を与えることがあるので,その取扱いや解析方法には注 意が必要である。すなわち,同一の測定条件下において得られた熱分析データを同一の方 法によりデータを解析した場合は,それらのEaを比較検討できると考えてよい。真空下, 大気圧下や雰囲気ガスなどの環境が異なる場合のデータを比較することは避ける方が安全 である。 1.7.4 昇温速度と反応速度 熱分析においては,一定の昇温速度β(dT/dt)(つまり時間に対して温度を直線的に上昇 させる)で試料を加熱し,データを取得することが多い。TPD は一定昇温速度で加熱する 方法であるから,TPD を用いるとこの方法による多くのデータを得ることができる。反応 速度の解析においても,この条件で得られたデータを用いて解析するものが多いので,そ れに関する式を以下に示す。 一定の昇温速度β(dT/dt)で試料を加熱すると,時間 t における試料の温度 T は T = T0 + β t (34) となる。熱分解の反応速度式((33)式)は以下のように書き直される。 − a dα dα A = = × e RT f (α ) dt β dT β E (35) 23 第1章 昇温脱離法および関連することがら この式の誘導は以下のとおりである。 T = T0 + β t より, dT = β dt → dt = 1 β dT (36) 反応速度式((23)式)にこれを代入する。 − a − a dα dα A = A × e RT f (α ) → = × e RT f (α ) 1 dT β dT E E (37) β 以上で(35)式が誘導された。 (35)式を変形する。 dα A − a = × e RT dT f (α ) β E (38) 時間T0(反応スタート時)において反応率はゼロであり,時間Tにおいて反応率をαとす る。時間T0からTまで積分する。積分形を得るため関数g(α)を導入する。簡単のためT0=0 としてもよい。 g (α ) = ∫ α 0 AEa dα A T − a = ∫ e RT dT = p (x ) T βR f (α ) β 0 E (39) ここで,右辺に導入されたp(x)関数は指数積分関数Eiを含む関数で,x=Ea/RTとして下式 で示される。なお,Eiも合わせて示す。 e− x − Ei (− x ) x −x ∞ e Ei (− x ) = − ∫ dx x x p(x ) = (40) (41) 24 第1章 昇温脱離法および関連することがら 1.7.5 解析方法―等温的方法(Isothermal kinetic analysis) 等温的方法は,一定温度で測定したいくつかの熱分析データを用いて解析する方法であ る。得られたデータからdα/dt(反応速度)を求める。微分法ではdα/dtに対していくつか のモデル関数を当てはめてプロットする。プロットの直線性や原点からの偏差を比較して 最も適合するモデル関数を決める。モデル関数が決まるとk(速度定数)が求められる。1/T に対するkのアレニウスプロットからEaを算出することができる。 この方法は,一定温度において試料を加熱するのだから,測定中に反応が完結しなかっ たり,測定前に反応が進行するという点が問題となる。だから,測定に用いられる温度領 域が狭くなる。また,適切なモデル関数を選択するのが困難な場合があるが,直線性の良 好なモデル関数が選択できればモデル関数によらず得られたEa値はよい一致性を示すと言 われている。 1.7.6 解析方法―非等温的方法(Non-isothermal kinetic analysis) 非等温的方法は,一定の速度で昇温して得られた熱分析カーブを用いる方法である。こ の方法はさらに分類され,単一測定法,等変化率法(isoconversion method),ピーク法お よびジャンプ法がある。 1)単一測定法 単一測定法はある昇温速度における一つの熱分析カーブで解析する方法である。Sharp とWentworthの方法,Coats-Redfernの方法,Freeman-Carrollの方法およびAndersonと Freemanの方法がある。この方法は解析上いくつかの問題点があり注意すべきであり,使 用を推奨しない報告もある31,32) 。 2)等変化率法 等変化率法は,いくつかの昇温速度で得られた一連の熱分析データのうち同一の反応率 の デ ー タ を 用 い る 方 法 で あ る 。 等 変 化 率 法 に は , 微 分 型 の Friedman 法 と 積 分 型 の Flynn-Wall-Ozawa(FWO)法33,34)*8がある。 Friedman 法は熱分解の反応速度式((33)式)を用いる。 − a dα = A × e RT f (α ) dt E (42) 両辺の自然対数をとると 8 Ozawa法ともいう。小沢先生の論文34)でFlynn-Wall-Ozawa(FWO)法と記されていたので,ここでは FWO法とする。 25 第1章 昇温脱離法および関連することがら ln E dα = ln [Af (α )] − a dt RT (43) が得られる。一定のαに対してはln[Af(α)]は定数となる。昇温速度を変えて得られたい くつかのデータからあるαに対するdα/dtと 1/Tの組みをプロットする。得られた直線の傾 きは,−Ea/RであるからここからEaを求める。 FWO法31,32)では,(39)式を用いるがp(x)関数として,Doyleによる近似式((44)式)を用 いている。Doyleは下で示すxが 20 以上のとき,p(x)は下式で近似できるとした。 log p( x ) = −2.315 − 0.4567 x (44) ここで,x=Ea/RTである。 反応速度の積分型を書き直すと(45)式となる。 g (α ) = AE a AE a AE a p(x ) → β = × p ( x ) → log β = log + log p ( x ) Rg (α ) Rg (α ) βR (45) Doyleの近似式を代入し,x=Ea/RTとすると(46)式が得られる。 log β = log AE a E − 2.315 − 0.4567 a Rg (α ) RT (46) αが一定の場合は右辺の対数項は定数となる。そうすると,反応が単一の機構で進行して いるのなら,いくつかのβに対して,一定のαのときのTを測定して,その逆数(1/T)とlog βをプロットすると,直線が得られる。その傾きMからEaは(47)式で示され,活性化エネル ギーEaを求めることができる。 Ea = − RM 0.4567 (47) 3)その他の方法 その他の方法として以下のものがある。 ピーク法は,いくつかの昇温速度で得られた一連の熱分析のデータのうち,反応速度が ピーク(最大)を示す温度を用いて解析する方法である。Kissinger 法が代表的である。 26 第1章 昇温脱離法および関連することがら 微分型の熱分解のデータ(TG,DTA,DSCカーブ,TPDカーブ)を用いる。反応速度式 を時間に対して微分する(d2α/dt2)と反応の最大値(カーブのピーク)では ⎛ d 2α ⎜⎜ 2 ⎝ dt ⎞ ⎟⎟ = 0 ⎠p であるから,(33)式より(48)式が得られる。 ln ⎡ ⎛ df (α ) ⎞ AR ⎤ E a 1 × = ln ⎢ − ⎜ ⎟ ⎥− T ⎢⎣ ⎝ dα ⎠ p E a ⎥⎦ R T p β (48) 2 p 右辺の対数項が定数になる場合には,1/Tpに対してlnβ/Tp2をプロットするとその傾きか ら活性化エネルギーを求めることができる。 ジャンプ法には,温度ジャンプ法と速度ジャンプ法がある。温度ジャンプ法は,等温的 測定の過程で設定温度を瞬間的に変化させ,そのときの反応速度の変化を測定するもので ある。速度ジャンプ法は,速度制御熱分解法において反応速度(制御反応速度)を瞬間的 に変化させて,そのときの温度変化を測定するものである。 等温的方法と非等温的方法で得られたEaは異なり,比較できないこともあるから注意す べきであるとの報告もある35)。 27 第1章 昇温脱離法および関連することがら 以上の方法により固体の熱分解における活性化エネルギーを求めることができる。 固体の熱分解反応における活性化エネルギーについて,一般的に言われている知見を述べ る36)。 1) 熱分解反応が不可逆反応の場合は,得られたEaは反応条件に依存せず,よい一致を示 すことが多い。 2) 可逆反応の場合は,測定機器の影響を受けやすい。(測定機器による反応条件の差異を 厳密にはなくすることはできない,ということ) 3) 反応条件により反応が影響を受ける場合には,調べた温度領域を明示して,その反応条 件下のみで有効である。多くの固体の熱分解反応はこれに属すると考えてよい。 それに加えて,以下のことも考慮しなければならない。固体の熱分解におけるEaは, 4) 活性化エネルギーを求める式により値が異なる。 5) 反応率(α)に依存して変化する。 6) 測定している雰囲気により値が異なる。 7) 測定方法により値が異なる。 8) 測定テクニックによっても異なる値を示す。 28 第1章 昇温脱離法および関連することがら 1.8 昇温脱離装置と TPD カーブ 1.8.1 昇温脱離装置 1)昇温脱離装置 図 1.11 に昇温脱離(TPD)装置の概念図を示す。試料室やロードロック室はターボ分子 ポンプ(TMP)で超高真空になっている。通常状態で試料室は 10−7∼10−8Paオーダーの高 真空に保持されている。 QMSは,試料の中心軸から傾斜されており(約 19°),試料表面から約 10cmの距離に配 置されている。これが,下方からの赤外線の悪影響を受けず,脱離ガスを最も効率良く検 出できる配置である。そのため,10−10モル以下の微量脱離ガスを測定することができる。 分析にあたっては,QMSの信号強度が 10−9A程度を上限とすると装置の真空度を低下させ ないで測定できる。この場合は 10−11A程度の強度を示すm/zまでが信頼のおける解析対象 であろう*9。 A nalysis cham ber Lo ad -lock cham ber V a cuum ga u ge Ionization ga uge QMS Sam p le stage Sam p le tra nsfer unit Sam p le tra nsfer u nit TM P RP IR heatin g u nit TM P T em p erature co ntroller RP C om p uter unit 図 1.11 TPD 装置の概念図 2)試料の測定 解析する試料は汚染されないように細心の注意をはらって作製すべきである。汚染物質 による TPD カーブを試料からのそれと誤認してしまうことのないようにすべきである。汚 染物質による試料室内の汚染の危険も避けねばならない。また,不必要な水分や溶媒を試 料から除去しておくべきである。これらの蒸発によりロードロック室や試料室の圧力がい つまでも下がらなければ測定が不可能になるからである。 9 この値はQMSの設定電圧で変化するので一応の目安である。 29 第1章 昇温脱離法および関連することがら 汚染物質の脱離には細心の注意を払って事前に調べておくべきである。特にFは試料室を 汚染しそれの除去は特殊な手段によらねばならないので,Fの脱離は事前に確認すべきであ る。また,金属が脱離するとそれがQMSを汚染し感度低下や故障の原因になるから,これ も事前に調べておくべきである。これらの物質の脱離が考えられる場合には,事前にすべ てのm/zについてQMS信号強度を測定する方法*10を用いて調べておく。この測定を行って いるときに汚染物質の脱離が少量でも観測された(またはその兆候がある)のなら,そこ でその測定は中止すべきである。その後の試料の測定時には,汚染物質の脱離前で測定を 終了するか測定そのものを中止すべきである。Fが脱離した場合は,測定後F除去剤を用い て除去作業を行う。 研究対象の脱離ガス種は事前に予測して測定するm/zを決める。それが困難な場合は,す べてのm/zについてまずQMS信号強度を測定して対象となるm/zを決定する*11。 試料はまずロードロック室に入れる。次に,これを試料室内のサンプルステージにマニ ピュレーターで移載する。試料は下方からのハロゲンランプによる赤外線で加熱される。 薄膜試料の場合,基板としてシリコンウェーハを用いると,基板のみが優先的に加熱され 装置内の加熱は抑えられる。その結果,主なガス発生は試料からのみとなり,データのバ ックグラウンドがきわめて低くなり解析が容易となる。 試料温度は試料台に設置した熱電対または試料表面にセットした熱電対でモニターされ る。温度のモニターは試料に接触した熱電対で行うのがよい。高温領域では,両者の解離 が大きくなりステージ温度の追跡では試料温度を正確に測定しているとは言えないからで ある。 昇温速度の選定は注意すべきである。昇温速度が試料の熱反応(熱分解)に対して遅す ぎると,試料室の器壁や脱離ガスの再吸着の影響により TPD カーブが本来のものから変化 する。昇温速度が速すぎると,試料が熱反応する前に温度が上昇し熱反応がまわりの温度 に追従しなくなるからである。一般的には 10∼20℃/min が適切であろう。また,脱離の活 性化エネルギーを求める場合には,解析式にもよるが昇温速度を 1∼10℃/min まで変化し て TPD カーブを測定する。 10 11 SCAN測定である。 SCAN測定である。 30 第1章 昇温脱離法および関連することがら 3)QMS によるガス分析 放出されたガスは次のようにして分析される。図 1.12 にその模式図を示す。高真空下で 試料を加熱すると,ある温度領域で試料から吸着物質が放出される。また,試料の表面お よびその近傍の物質には,加熱によりガスとして放出されるものがある。ガスはある脱離 速度で放出され,装置内の圧力が変化する。放出されたガスは,QMS によってそれぞれの 質量電荷数(,m/z)に対応したデータを示す。加熱には種々のパターンがあるが,一定速度 の昇温速度とすると取り扱いやすい。従って,加熱温度(加熱時間)に対して,脱離ガス の m/z に対応した QMS 強度は変化する。これを TPD カーブ(曲線) (TPD curve)とい う。 「スペクトル」は,狭義では波長などの光学的尺 (TPD スペクトルや TDS スペクトルという表記もある。 度などをとって縦軸にそれに対するある変化を示すものである。熱分析では横軸に温度をとり重量変化な どを示す。このときはたとえば TG カーブといい「カーブ」という表記を用いる。だから,本書では TPD カーブという呼称を用いる。しかし,スペクトルは,広義では横軸にエネルギーまたはそれに相当する量 をとって,それに対する物性などの変化を示すという定義があるから,それに基づくと「スペクトル」と いう表記もなりたつ。また,TPD は TDS(Thermal Desorption Spectroscopy)ともいうので,TDS スペ クトルや TPD スペクトルという呼称もあるのは理解できる。論文では両者が用いられているが,「スペク トル」が多い。) QMS 脱離ガスを質量分析計で測定 TPDカーブ 真空中 (10-7∼10-8Pa) 脱離ガス 赤外線 図1.12 昇温脱離法の概念図 31 第1章 昇温脱離法および関連することがら 1.8.2 TPD カーブ TPD カーブの例を以下に示す。TPD カーブの形状や解析法については,第 2 章で詳しく 述べる。 1)チタニアゲル膜に物理吸着したH2OのTPDカーブ 例として図 1.13 にチタニアゲル膜に物理吸着した水のTPDカーブを示す(筆者らのデー タ 未発表) 。脱離したH2O(m/z 18*12*13)が,横軸を温度,縦軸をQMS強度として示さ れる。この図では,H2Oは 50℃から 150℃の温度領域で脱離していることが分かる。脱離 の温度領域やカーブの形状からH2Oの状態(物理吸着や化学吸着,試料の細孔内に吸着して いるなど)を解析できる。また,脱離量はカーブの面積に対応しており,カーブの面積を 求め標準試料と比較検討することによりそれは求められる。この例ではピークトップ温度 が 100℃以下と低いので物理吸着であると判断された。また,そのカーブ形状より吸着状態 強度 は単一であろうと考えられる。 100 200 300 400 500 温度 / ℃ 図1.13 H2O(m/z 18)のTPDカーブ 2)α-Cr2O3(001)結晶に吸着したH2OのTPDカーブ TPDカーブの例をもう一つ示す。図 1.14 は,α-Cr2O3(001)結晶に 120KでH2O蒸気にさ 12 QMS内に入ったガスはイオン(カチオン)化されるので,その物質は質量電荷比(m/zまたはm/e) (m: 質量z,e:電荷数)であらわされる。 13 H2Oは,自然界に最も多く存在している同位体1Hと16Oで構成されていると考えている。 32 第1章 昇温脱離法および関連することがら らし,結晶に吸着したH2O(m/z 18)のTPDカーブである37)。まず,H2Oの表面濃度が低 いとき 345K(72℃)にピークを示すカーブが観測され,9∼10×1014molecules/cm2で飽和 した。次に 295K(22℃)にピークが見られるが,14×1014molecules/cm2で飽和した。つ いで,210K(−63℃)と 185K(−88℃)にピークが観測され,20×1014molecules/cm2で 飽和した。この飽和に対応して 165Kにピークが観測された。345Kと 210Kのカーブは化学 吸着水に対応しており,それ以下のカーブは物理吸着水に対応している。このカーブの変 化は吸着水の層構造によっている。すなわち,H2Oはまず化学吸着し(345Kのカーブに対 応)次に同様に 295Kのカーブに対応した化学吸着する。ついで,物理吸着していくが,よ り低温のカーブは上層のH2Oに対応しているのである。 3)α-Fe2O3(012)表面に吸着したH2OのTPDカーブ 擬似的な 0 次脱離と 1 次脱離のTPDカーブを次に示す。図 1.15 はα-Fe2O3(012)の(2 ×1)表面を 120KでH2O蒸気にさらし,結晶に吸着したH2O(m/z 18)のTPDカーブであ る38)。表面濃度を変えてTPDカーブを測定した。400K付近に観測されるTPDカーブは濃度 の増大とともにピークが高温側へシフトしており擬似的な 0 次の脱離を示している。 図 1.16 はα-Fe2O3(012)の(1×1)表面を 120KでH2O蒸気にさらし,結晶に吸着した H2O(m/z 18)のTPDカーブである38) 。表面濃度を変えてTPDカーブを測定した。表面濃 度が低いとき 350Kにピークが観測され,濃度の増大とともにピーク強度が大きくなった。 濃度増加によってもピーク温度は変化せず,またその形状からこの脱離は 1 次の脱離であ る。 33 第1章 昇温脱離法および関連することがら 図 1.14 α-Cr2O3(001)結晶に吸着したH2O(m/z 18)のTPDカーブ37) 34 第1章 昇温脱離法および関連することがら 図 1.15 α-Fe2O3(012)の(2×1)表面に吸着したH2OのTPDカーブ38) 図 1.16 α-Fe2O3(012)の(1×1)表面に吸着したH2OのTPDカーブ38) 35 第1章 昇温脱離法および関連することがら 文献 1. R. I. Masel, “Principles of Adsorption and Reaction on Solid Surfaces”, John Wiley & Sons, NY, (1996). 2. J. B. Hudson, “Surface Science an Introduction”, John Willey & sons, New York, 1992, pp. 220-225. 3. H. Lüth,”Solid Surface, Interfaces and Thin Films”, Springer, Berlin, 2001, pp. 526-533. 4. J. W. Niemantsverdriet, “Spectroscopy in Catalysis, an Introduction”, 2nd ed., Wiley-VCH, Weinheim, 2000, pp.22-36. 5. R. J. Cvetanović and Y. Amemiya, Advan. Catal., 17 (1967) 103-149. 6. R. J. Cvetanović and Y. Amemiya, Catal. Rev., 6 (1972) 21-48. 7. M. Benjaram, M. P. Sreekanth, R. V. Pavani, Journal of Molecular Catalysis A: Chemical, 225 (2005) 71-78. 8. D. Tsiplakides, S. Balomenou, A. Katsaounis, D. Archonta, C. Koutsodontis and C. G. Vayenas, Catal. Today, 100 (2005) 133-144. 9. A. Tungler, J. Thermal Anal. Calor., 79 (2005) 521-524. 10. 平下紀夫,内山泰三,分析化学,43 (1994) 757-763. 11. Y. Miyakawa, J. Hashimoto, N. Ikegami, T. Matsui and J. Kanamori, Jpn. J. Appl. Phys., 33 (1994) 2145-2150. 12. 藪本周邦,表面技術,46 (1995) 249-252. 13. T. Feng and Xu Guo Qin, Accounts Chem. Res., 37 (2004) 882-893. 14. G. Spoto, E. N. Gribov, G. Ricchiardi, A. Damin, D. Scarano, S. Bordiga, C. Lamberti and A. Zecchina, Prog. Surf. Sci., 76 (2004) 71-146. 15. M. Niwa and N. Katada, Hyomen Kagaku, 24 (2003) 635-641. 16. N Katada and M. Niwa, Zeoraito, 21 (2004) 45-52. 17. P. Acharya and G. H. Hay, Proceedings of the International Conference on Incineration and Thermal Treatment Technologies, (2000) 588-595. 18. M.Kawamoto, K. Ishizaki and C. ishizaki, “Handbook on characterization technique for the solid-solution interface”, Edited by J. H. Adair, J. A. Casey and S. Venigalla, The American Ceramic Society, Ohio, 1993, pp.283-292. 19. 中柗哲也,石崎幸三,セラミックス,32 (1997) 646-650. 20. 津越敬寿,セラミックス,37 (2002) 81-83. 21. T. Nishide, T. Yabe, N. Miyabayashi and Makiko Sano Thin Solid Films, 467 (2004) 43-49. 22. T. Takahashi and T. Nishide, J. Ceram. Soc.Jpn. 112 (2004) S234-S238. 23. T. Nishide, T. Meguro, S. Suzuki and T. Yabe, J. Ceram. Soc.Jpn. 113 (2005) 77-81. 36 第1章 24. 昇温脱離法および関連することがら 新訂版・表面科学の基礎と応用 ,日本表面科学会編,エヌ・ティー・エス,2004, p. 361,369. 25. 新訂版・表面科学の基礎と応用”,日本表面科学会編,エヌ・ティー・エス,p. 64. 26. J. B. Hudson, “Surface Science, an Introduction”, John Willy & Sons, New York, 1998, p. 200. 27. C. H. Bamford and C. F. H. Tipper ed. “Chemical Kinetics”, Elsevier, Amsterdam 1980, p.42. 28. J. Sesták, and G. Berggren, Thermochimica Acta, 3 (1971) 1-12. 29. J. Málek and J. M. Criado, Thermochimica Acta, 175 (1991) 305-309. 30. H. L. Anderson, A. Kemmler, G. W. H. Höhne, K. Heldt and R. Strey, Thermochimica Acta, 332 (1999) 33-53. 31. M. Maciejewski, Thermochimica Acta, 355 (2000) 145-154. 32. A. K. Burnham, Thermochimica acta, 255 (2000) 165-170. 33. T. Ozawa, Bull. Chem. Soc. Jpn., 38 (1965) 1881-1886. 34. T. Ozawa, Thermochimica Acta, 203 (1992) 159-165 35. B. Roduit, Thermochimica Acta, 355 (2000) 171-180. 36. A. K. Galwey and M. E. Brown, “Thermal Decomposition of Ionic Solids”, (Studies in Physical and Theoretical Chemistry 86) Elsevier, Amsterdam (1999) p. 122.) 37. M. A. Henderson and S. Chambers, Surf. Sci., 449 (2000) 135-150. 38. M. A. Henderson, S. A. Joyce and J. R. Rustad, Surf. Sci, 417 (1998) 66-81. 37
© Copyright 2026 Paperzz