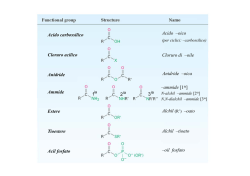
ACIDI CARBOSSILICI Molti acidi carbossilici sono di origine naturale e hanno un odore e un sapore caratteristico. Ac. formico Ac. acetico Ac. butirrico Ac. Ossalico HOOC-COOH L’acido formico, che ha un odore acre e un gusto “mordente”, viene secreto dal pungiglione di alcuni tipi di formiche. Il nome è derivato proprio dalla parola latina formica. L’acido acetico è il componente acido dell’aceto. Il nome viene proprio dal latino acetum. L’ossidazione aerobica dell’etanolo ad acido acetico è il processo che contraddistingue il decadimento del vino cattivo. L’acido acetico è una materia prima industriale per la produzione di polimeri per vernici e per adesivi. L’acido butanoico è un prodotto di ossidazione che contribuisce all’odore sgradevole di un corpo sudato. Il suo nome comune, acido butirrico, deriva dal latino butyrum che significa burro, perché l’acido butirrico dà l’odore e il sapore peculiare al burro rancido. L’acido ossalico si trova in natura negli spinaci e nel rabarbaro. Sebbene sia tossico, si dovrebbero mangiare quattro chili e mezzo di spinaci perché se ne ingerisca una dose letale. H O H H C H C C O H O H Acido lattico L’acido lattico dà al latte acido il suo sapore inequivocabile. L’acido lattico si forma anche nel metabolismo del glucosio a CO2 e H2O. Durante l’attività fisica l’acido lattico si forma a una velocità maggiore rispetto a quella della sua ulteriore ossidazione, e questo dà luogo alla sensazione dolorosa di muscoli stanchi. Quando esso viene metabolizzato questa sensazione scompare. Molti acidi carbossilici hanno nomi comuni utilizzati più frequentemente dei loro nomi IUPAC. Oltre il C5 gli acidi carbossilici sono acidi naturali di formula CH3(CH2)nCOOH con n pari. REAZIONI DEGLI ACIDI CARBOSSILICI E DERIVATI Come è già stato detto, la reazione caratteristica degli acidi carbossilici e dei suoi derivati è la reazione di sostituzione nucleofila. Questa è una reazione generale che avviene sia con nucleofili carichi negativamente, sia con nucleofili neutri (talvolta con catalisi acida o basica). O O - R C Z :Nu o HNu: R C - Nu + Z: o H-Z gruppo uscente Il meccanismo generale per le reazioni di sostituzione nucleofila acilica prevede un processo a due stadi: Nello stadio 1 il nucleofilo attacca il gruppo carbonilico, promuovendo la rottura del legame e formando un nuovo legame C-Nu. Nello stadio 2, l’eliminazione del gruppo uscente ed il contemporaneo ripristino del legame C-O forma il prodotto di sostituzione. La sostituzione nucleofila avviene con carbanioni (:R-), idruri (:H-) e con una vasta gamma di nucleofili all’ossigeno e all’azoto: Nucleofili all’azoto Nucleofili all’ossigeno - O OH H2O ROH R C O NH3 - RNH2 R2NH Gli acidi carbossilici e i loro derivati differiscono notevolmente nella reattività nei riguardi dei nucleofili. L’ordine di reattività segue la facilità di uscita del gruppo uscente. Facilità di eliminazione crescente Gruppo uscente - R NH2 - OR - OH RCOO- Cl- O O O O O C NH2 R C OR R C OH R C O O C R R C Cl Reattività crescente I composti acilici più reattivi possono essere convertiti in quelli meno reattivi. La reazione inversa è di solito molto più difficoltosa. Per spiegare questo comportamento bisogna ricordare che il nucleofilo si addiziona al carbonile formando un intermedio tetraedrico che contiene due potenziali gruppi uscenti, o :Nu- o :Z-. Il gruppo che verrà successivamente eliminato è quello, tra questi due, che uscirà più facilmente, cioè il miglior gruppo uscente. Perché la reazione avvenga, Z deve essere un miglior gruppo uscente rispetto a Nu I due possibili gruppi uscenti La sostituzione nucleofila avviene quando un gruppo uscente :Z- è una base più debole e quindi un miglior gruppo uscente rispetto al nucleofilo :Nu- che attacca. RIDUZIONE DI ACIDI, ESTERI E CLORURI ACILICI La riduzione degli acidi carbossilici e dei suoi derivati porta a prodotti differenti a seconda del riducente utilizzato. Gli idruri metallici sono i riducenti più utili. L’idruro di litio e alluminio è un riducente forte e reagisce con tutti i composti carbonilici. Esso converte acidi, esteri e cloruri acilici in alcoli primari. O CH3CH2CH2 C OH 1) LiAlH4 2) H2O CH3CH2CH2CH2OH + H2O OCH3 1) LiAlH4 2) H2O CH3CH2CH2CH2OH + CH3OH 1) LiAlH4 2) H2O CH3CH2CH2OH + HCl O CH3CH2CH2 C O CH3CH2 C Cl Ad un’iniziale sostituzione nucleofila segue un’addizione nucleofila sull’aldeide che si forma nella prima parte del processo. L’attacco nucleofilo dello ione idruro sul carbonio del gruppo carbonilico forma un intermedio tetraedrico di natura alcossidica. L’ossigeno carico negativamente utilizza uno dei suoi doppietti elettronici non condivisi per ripristinare il legame C-O, contemporaneamente si rompe il legame C-Z con espulsione di :Z- e formazione di un’aldeide. Un altro ione idruro attacca il carbonile dell’aldeide che si è formata originando, secondo il classico meccanismo di addizione nucleofila al carbonile, uno ione alcossido che dopo protonazione porta all’alcool primario. Nella riduzione di un acido carbossilico, lo ione idrossido si comporta da gruppo uscente e viene poi protonato a formare acqua; nel caso di un estere il gruppo uscente è il gruppo alcossido che viene poi protonato a formare un’altra molecola di alcol oltre a quella ottenuta per riduzione. Nella riduzione di un cloruro acilico, lo ione cloruro è il gruppo uscente e viene poi protonato ad acido cloridrico. Riducenti più deboli come l’idruro di diisobutilalluminio (DIBAH o DIBAL-H) e l’idruro di litio tri-t-butossialluminio, trasformano rispettivamente a bassa temperatura esteri e cloruri acilici in aldeidi, ma non sono in grado di attaccare gli acidi carbossilici. In questo caso il processo si ferma dopo la reazione di un equivalente di ione idruro. Il DIBAH ha due voluminosi gruppi isobutile che rendono questo reagente meno reattivo dell’idruro di litio e alluminio. L’idruro di litio tri-t-butossialluminio, ha tre atomi di ossigeno elettronegativi che lo rendono meno nucleofilo dell’idruro di litio e alluminio. OC(CH3)3 Al H = [CH3)2CHCH2]2AlH Li+ H Al OC(CH3)3 = LiAlH[OC(CH3)3]3 OC(CH3)3 DIBAH idruro di litio tri-t-butossialluminio REAZIONI DEI CLORURI DEGLI ACIDI I cloruri acilici reagiscono con nucleofili per formare prodotti di sostituzione. Poiché durante tali reazioni si forma acido cloridrico che costituisce un sottoprodotto indesiderato, si lavora con un eccesso di nucleofilo/base o, in caso questo fosse troppo costoso, aggiungendo un’altra base per neutralizzarlo. O R C O Cl + H Nu: R C Nu + H Cl :B + - B-H Cl Reagendo con nucleofili all’ossigeno, dai cloruri acilici si ottengono anidridi, acidi carbossilici ed esteri. Reagendo con ammoniaca, ammine primarie e secondarie, i cloruri acilici si convertono in ammidi. Con nucleofili come gli ioni carbossilato, il meccanismo seguito è quello generale a due stadi: attacco nucleofilo seguito dalla fuoriuscita del gruppo uscente. 1. L’atomo di ossigeno carico negativamente dell’anione acetato utilizza uno dei suoi doppietti elettronici non condivisi per formare un nuovo legame con l’atomo di carbonio del gruppo carbonilico, contemporaneamente si rompe il legame carbonioossigeno, gli elettroni del legame vengono attratti dall’ossigeno, elemento più elettronegativo. Si ottiene un intermedio tetraedrico di natura alcossidica. 2. L’ossigeno carico negativamente utilizza uno dei suoi doppietti elettronici non condivisi per ripristinare il legame C-O, contemporaneamente si rompe il legame C-Cl con espulsione di uno ione cloruro e formazione di un’anidride. Con questo metodo è possibile preparare tanto le anidridi simmetriche quanto quelle miste. La sostituzione nucleofila con un nucleofilo neutro richiede uno stadio addizionale per il trasferimento di un protone: Idrolisi di un cloruro acilico Alcolisi di un cloruro acilico Ammonolisi di un cloruro acilico 1. Il nucleofilo (H2O, ROH, NH3) utilizza uno dei suoi doppietti elettronici non condivisi per formare un nuovo legame con l’atomo di carbonio del gruppo carbonilico, contemporaneamente si rompe il legame carbonio-ossigeno, gli elettroni del legame vengono attratti dall’ossigeno, elemento più elettronegativo. Si ottiene uno ione dipolare tetraedrico perché il carbonio del vecchio gruppo carbonilico muta ibridazione da sp2 a sp3. 2. L’ossigeno carico negativamente utilizza uno dei suoi doppietti elettronici non condivisi per ripristinare il legame C-O, contemporaneamente si rompe il legame C-Y con espulsione di uno ione alogenuro e formazione di un derivato di un acido carbossilico o di un acido carbossilico protonati. 3. Una base (anche il gruppo uscente) strappa il protone con conseguente liberazione del prodotto di sostituzione neutro. REAZIONI DELLE ANIDRIDI Le anidridi sono meno reattive dei cloruri acilici, ciò nonostante esse reagiscono facilmente con la maggior parte dei nucleofili per formare dei prodotti di sostituzione. La sostituzione nucleofila avviene a uno dei due gruppi carbonilici, mentre il secondo gruppo carbonilico diventa parte del gruppo uscente dando origine ad uno ione carbossilato. Quindi soltanto la metà della molecola viene utilizzata. O R C O O C O R + H Nu: R C O Nu + HO C R sottoprodotto Nu sostituisce RCOO Le anidridi non possono essere convertite in cloruri acilici, perché RCOO- è una base più forte e quindi un gruppo uscente peggiore dello ione Cl-. Le anidridi possono, tuttavia, essere utilizzate per preparare tutti gli altri derivati acilici. In queste reazioni viene sempre ottenuta come sottoprodotto una molecola di acido carbossilico o di un suo sale. Idrolisi R O O C C O O R + H2O R C O OH + HO C R sottoprodotto Alcolisi O R C O O C O R + R'OH R C O OR' + HO C R sottoprodotto Ammonolisi R O O C C O O R + 2NH3 R C O NH2 + C + NH4 O sottoprodotto R L’ammonolisi degli esteri è una reazione scarsamente utilizzata, perché normalmente è più facile partire da un cloruro acilico. Il meccanismo di conversione di un’anidride in acido carbossilico, estere o ammide è identico a quello delle analoghe reazioni dei cloruri acilici e avviene in tre stadi: addizione nucleofila, eliminazione del gruppo uscente e deprotonazione del prodotto. R O O C C O O R - R C O O O C C R R O Nu + H - O C R H Nu H Nu H-Nu = H-OH, RO-H O R R C O O C C O Nu H + - O O O C C R OR R C O R Nu O O C C R R + O C NH3 H O N H H + :NH3 R C NH2 + HO C R O NH3 NH3 R O +NH 4 + - O C R 1. Il nucleofilo (H2O, ROH, NH3) utilizza uno dei suoi doppietti elettronici non condivisi per formare un nuovo legame con uno degli atomi di carbonio dei gruppi carbonilici, contemporaneamente si rompe il legame carbonio-ossigeno, gli elettroni del legame vengono attratti dall’ossigeno, elemento più elettronegativo. Si ottiene uno ione dipolare tetraedrico perché il carbonio del vecchio gruppo carbonilico muta ibridazione da sp2 a sp3. 2. L’ossigeno carico negativamente utilizza uno dei suoi doppietti elettronici non condivisi per ripristinare il legame C-O, contemporaneamente si rompe il legame C-OCOR con espulsione di uno ione carbossilato. 3. Lo ione carbossilato o una base strappa il protone dal prodotto di sostituzione protonato con conseguente liberazione del prodotto neutro. Le reazioni di alcolisi e ammonolisi dell’anidride acetica vengono sfruttate industrialmente per l’ottenimento di farmaci di uso comune. REAZIONI DEGLI ACIDI CARBOSSILICI Gli acidi carbossilici, presentando un legame polare O-H, danno reazioni acido-base veloci, e ogni nucleofilo che sia anche una base forte reagirà con l’acido rimuovendo un protone, prima che qualsiasi reazione di sostituzione nucleofila possa avvenire. O R C O O H + - :Nu R C O- + H-Nu Ciò nonostante, gli acidi carbossilici possono essere convertiti in una grande varietà di altri derivati acilici utilizzando dei reagenti particolari o attraverso una catalisi acida. CONVERSIONE DEGLI ACIDI CARBOSSILICI NEI CLORURI ACILICI Gli acidi carbossilici vengono trasformati più frequentemente nei loro cloruri che in qualsiasi altro derivato funzionale, perché i cloruri sono molto reattivi e da essi possono essere ottenuti molti altri composti. I cloruri acilici si preparano per sostituzione del gruppo ossidrilico dell’acido carbossilico con cloro. A tale scopo non si può utilizzare lo ione cloruro come nucleofilo in quanto è una base più debole del gruppo uscente, lo ione idrossido. Gli acidi carbossilici possono essere convertiti in cloruri acilici utilizzando tre reattivi, ma quello più conveniente è il cloruro di tionile perché i prodotti che si formano, oltre il cloruro dell’acido, sono dei gas che si separano facilmente. CONVERSIONE DEGLI ACIDI CARBOSSILICI IN ESTERI Un acido carbossilico viene trasformato direttamente in un estere per riscaldamento con un alcol in presenza di un catalizzatore acido. Questa reazione è reversibile e normalmente si raggiunge l’equilibrio quando, oltre ai prodotti di reazione, vi sono ancora sensibili quantità dei reagenti. Esterificazione di Fischer Nell’esterificazione di Fischer, l’equilibrio è particolarmente sfavorevole quando si abbia un gruppo stericamente voluminoso nelle vicinanze del centro reattivo. REATTIVITÀ NELL’ESTERIFICAZIONE CH3OH > 1° > 2° > 3° HCOOH > CH3COOH > RCH2COOH > R2CHCOOH > R3CCOOH Poiché la reazione è catalizzata da acidi, oltre ai due stadi usuali di addizione di un nucleofilo seguita dall’eliminazione di un gruppo uscente, ci sono due stadi addizionali di protonazione e deprotonazione. MECCANISMO DELL’ESTERIFICAZIONE DI FISCHER H Cl O R O R C H OH O H R' C O R OH O C H O OH R H R C O HO H O R' H R C OR' HO H C H OH OH2 O R C OR' H3O + 1. La protonazione del gruppo carbonilico dell’acido carbossilico ad opera di un acido lo rende più elettrofilo. 2. Una molecola di alcol attacca il carbonile protonato a dare un intermedio tetraedrico di natura cationica. 3. Il trasferimento di un protone tramite una reazione acido-base intramolecolare trasforma il gruppo -OH in un buon gruppo uscente. 4. L’espulsione di una molecola di acqua e la contestuale (o successiva) deprotonazione del secondo gruppo ossidrilico fornisce un estere. L’esterificazione di un acido carbossilico avviene in presenza di un acido ma non in presenza di una base. La base rimuove un protone dall’acido carbossilico formando un anione carbossilato che non reagisce con un nucleofilo elettron-ricco. Entrambe le specie sono elettron-ricche O R C O O-H + - :OH R C O - + H2O - OH agisce come base, non come nucleofilo O R'OH R C OR' ESTERI Aromatizzanti Gli aromatizzanti costituiscono la classe più numerosa degli additivi alimentari. Attualmente è disponibile più di un migliaio di aromi, tra naturali e sintetici. La maggioranza di questi viene concentrata o estratta da sostanze naturali. Gli aromatizzanti sono spesso miscele complesse formate da dieci fino a centinaia di composti. Un gran numero di agenti aromatizzanti, la maggioranza dei quali sono esteri, sono invece sintetizzati industrialmente. Molti di questi hanno aromi molto somiglianti a quelli richiesti e l’aggiunta di uno o pochi di essi è sufficiente per fare gelati, bevande analcoliche o caramelle con gusti naturali. Nome Formiato di etile Struttura HCOOCH2CH3 Aroma Rum Acetato di isopentile CH3COOCH2CH2CH(CH3)2 Banana Acetato di ottile CH3COOCH2(CH2)6CH3 Arancia Butanoato di metile CH3CH2CH2COOCH3 Mela Butanoato di etile CH3CH2CH2COOCH2CH3 Ananas Gli esteri reagiscono con ammoniaca e ammine primarie e secondarie per formare ammidi. L’ammonolisi degli esteri è tuttavia una reazione scarsamente utilizzata, perché normalmente è più facile partire da un cloruro acilico. Il meccanismo è perfettamente identico a quello dell’ammonolisi di un cloruro acilico o di un’anidride, cambia solo il gruppo uscente. L’ammoniaca attacca l’atomo di carbonio del gruppo carbonilico, contemporaneamente si rompe il legame carbonio-ossigeno, gli elettroni del legame vengono attratti dall’ossigeno, elemento più elettronegativo. Si ottiene uno ione dipolare tetraedrico. L’ossigeno alcossidico utilizza uno dei suoi doppietti elettronici non condivisi per ripristinare il legame C-O, contemporaneamente si rompe il legame C-OR con espulsione di uno ione alcossido, e formazione di un’ammide protonata sul gruppo amminico che viene deprotonata dallo ione alcossido con conseguente liberazione dell’ammide neutra. Idrolisi acida delle ammidi e degli esteri 1. Protonazione del carbonile ammidico o estereo ad opera di uno ione H3O+. 2. Una molecola d’acqua attacca il carbonile protonato a dare un intermedio tetraedrico. 3. Trasferimento di un protone che trasforma il gruppo -NH2 o il gruppo –OCH3 in un buon gruppo uscente. 4. L’espulsione della molecola di ammoniaca o di una molecola di metanolo fornisce un acido carbossilico protonato che viene deprotonato dalla stessa ammoniaca o da una molecola di acqua. Idrolisi basica delle ammidi e degli esteri (saponificazione) 1. L’addizione nucleofila dello ione ossidrile al carbonio carbonilico fornisce il consueto intermedio tetraedrico a carattere di alcossido. 2. L’eliminazione dello ione ammiduro o di uno ione alcossido genera l’acido carbossilico. 3. Lo ione ammiduro e lo ione alcossido, basi fortissime e cattivissimi gruppi uscenti, strappano, in maniera irreversibile, il protone acido dall’acido carbossilico e forniscono uno ione carbossilato e ammoniaca (o un’ammina primaria o secondaria) o alcol. I saponi I saponi sono conosciuti almeno sin dal 600 a.C., quando i Fenici preparavano una sostanza simile al latte cagliato, facendo bollire grasso di capra con estratti di cenere di legna. Le proprietà detergenti dei saponi non erano tuttavia riconosciute da tutti e l’uso del sapone non si diffuse fino al XVIII secolo. Chimicamente il sapone è una miscela di sali di sodio e potassio di acidi grassi a catena lunga prodotti per idrolisi di grassi animali (i triacilgliceroli) con alcali. La cenere della combustione del legno fu usata come sorgente di alcali fin dai primi dell’800, quando lo sviluppo del processo LeBlanc rese disponibile a livello commerciale Na2CO3. I saponi grezzi contengono glicerolo, gli alcali in eccesso e, ovviamente, sapone, ma possono essere purificati facendoli bollire con acqua e aggiungendo NaCl o KCl per precipitare i sali degli acidi carbossilici puri. Il sapone liscio che precipita viene seccato, profumato e pressato in pezzi per uso domestico. I saponi colorati si ottengono per aggiunta di un colorante, quelli per uso sanitario per aggiunta di sostanze antisettiche e quelli abrasivi per aggiunta di pomice. Se si desidera un sapone che galleggi ci si soffia dentro aria per diminuirne la densità. A parte tutti questi trattamenti extra e i costi che ne risultano, i saponi sono fondamentalmente tutti uguali. I saponi esercitano una funzione detergente perché le due estremità della molecola di sapone sono profondamente diverse tra loro. La parte carbossilata anionica della lunga catena molecolare è ionica e perciò idrofila; di conseguenza si scioglie in acqua. La lunga parte idrocarburica della molecola, invece, è apolare ed è attratta dal grasso. Il vantaggio di queste due opposte affinità è che i saponi sono attratti sia dai grassi che dall’acqua e sono perciò dei validi detergenti. Quando i saponi si disperdono in acqua, le lunghe code idrocarburiche delle molecole si raggruppano assieme formando l’interno idrofobico di una sorta di palla aggrovigliata, la micella, mentre le teste ioniche delle molecole si distribuiscono sulla superficie di questo aggregato a contatto con la soluzione acquosa. Le goccioline di olio e grasso diventano solubili in acqua perché vengono circondate dalle code apolari delle molecole di sapone e trattenute al centro delle micelle. Una volta resi solubili, il grasso e lo sporco possono essere sciacquati via. I saponi presentano molti vantaggi nella vita di ogni giorno, ma c’è anche il rovescio della medaglia. Nell’acqua dura, che contiene ioni metallici, i carbossilati di sodio solubili si trasformano in sali insolubili di calcio e magnesio, lasciando il familiare anello di schiuma sui bordi della vasca da bagno e la sfumatura grigia nei vestiti bianchi. I chimici hanno ovviato a questi problemi sintetizzando un gruppo di detergenti sintetici a base di sali a lunga catena idrocarburica di acidi alchilbenzensolfonici. Il principio di azione dei detergenti sintetici è lo stesso dei saponi. La coda della molecola di alchilbenzene è attratta dal grasso, mentre la testa anionica solfonata è attratta dall’acqua. Diversamente dai saponi, però, i detergenti solfonati non formano sali metallici insolubili nell’acqua dura e non lasciano perciò gli spiacevoli residui insolubili.
© Copyright 2026 Paperzz