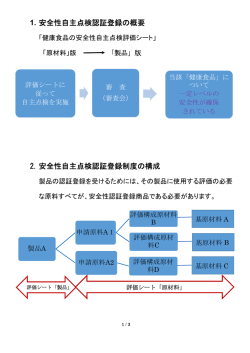
事 務 連 絡 平成 21 年 5 月 26 日 日本製薬団体連合会 御中 独立行政法人医薬品医療機器総合機構 品質管理部 定期調査に係る医薬品適合性調査申請時の留意点について 平成 17 年 4 月 1 日に施行した薬事法及び採血及び供血あつせん業取締法の一部を改正す る法律(平成 14 年法律第 96 号、以下、「改正薬事法」という。 )のみなし期間の経過措置 については、同法附則第 8 条に規定されているところですが、平成 22 年 3 月末まで製造販 売業者が製造販売承認を取得した品目のすべての経過措置期間が満了することから、今後、 医薬品医療機器総合機構(以下、「総合機構」という。 )に対する定期調査に係る医薬品適 合性調査申請が集中することが予想されます。つきましては、下記調査申請時の留意点に 御配慮いただくなど関係各位のご協力をお願いいたします。 記 1.医薬品適合性調査申請時期について 総合機構の医薬品適合性調査に要する標準的事務処理期間は 6 ヶ月とされており、定期 調査においてもみなし期間の経過措置が切れる 6 ヶ月前に調査申請するようお願いしてい るところではありますが、調査申請が一度に集中することも踏まえ、可能な限り早い時期 に申請をお願いいたします。なお、平成 21 年 10 月 1 日以降の調査申請については、平成 22 年 3 月末までに調査結果を通知できないこともありますのでご承知おきください。 2.医薬品適合性調査申請時の添付資料について (1)調査申請時に添付する資料としては、平成 17 年 3 月 30 日付薬食監麻発第 0330001 号厚生労働省医薬食品局監視指導・麻薬対策課長通知「薬事法及び採血及び供血あつ せん業取締法の一部を改正する法律の施行に伴う医薬品、医療機器等の製造管理及び 品質管理(GMP/QMS)に係る省令及び告示の制定及び改廃について」 (以下、 「G MP課長通知」という。 ) 第 3 9. (2)に示すほか、別添 1 に示すGMP調査用資 料を添付していただきますようにお願いいたします。なお、別添1の資料が添付され ていない場合でも医薬品適合性調査申請書の申請は可能ですが、調査申請後に調査員 から当該資料の請求に係る照会をすることになりますので、早急に準備を進め、提出 をお願いいたします。 (2)平成 16 年 9 月 22 日付厚生労働省令第 136 号「医薬品、医薬部外品、化粧品及び医 療機器の品質管理の基準に関する省令」の第 10 条の規定に基づき、総合機構の調査対 象となる製造所のGMPの適合性について、製造販売業者自らが実地で確認したこと を証する「製造販売業者によるGMP適合性等の確認書」 (以下、「確認書」という。 ) の添付にご協力をお願いいたします。提出する確認書には、製造販売業者の総括製造 販売責任者の署名あるいは記名押印をお願いします。調査申請時に確認書が添付され ない場合でも申請は可能です。 なお、製造販売業者自らが実地で確認できない場合には、他者の確認結果を利用す ることもできますが、その場合は、製造販売業者が他者の確認結果を確認し、総括製 造販売責任者が署名あるいは記名押印のうえ、当該確認書を提出してください。ご参 考までに別添 2 に確認書の例を添付しますので必要に応じ、活用してください。 (3)製造所が複数の品目を製造している場合、代表とする品目について、GMP課長通 知に従い、明白な根拠をもって選定したうえで、別添 1 についての資料提出をお願い いたします。 (4)調査対象品目について、改正薬事法に適合した内容となるよう承認書の記載整備を 行うこととされていますが、記載整備を行うべき期限が終了していない製造販売業者に おかれましては、承認書の記載整備の案の提出をお願いします。 3.その他 (1)定期調査に係る医薬品適合性調査は、製造品目、製造工程の難しさ、製造所の状況 等の情報のほか、上記提出資料内容を加味して書面による調査か実地による調査かの 判断を行います。 (2)書面による調査と判断された申請において、別添 1 の調査資料の提出や調査員から の照会事項への対応が滞っている場合は早急に品質管理部に連絡をお願いいたします。 (3)本取り扱いは、改正薬事法の施行に伴う当分の間の対応であり、平成 22 年 3 月末以 降は、状況に応じて適宜見直す予定としています。 別添1 ○定期調査に必要とする資料 1. 製造所の概要及び品目の概要に関する書類 2. 製造所の配置図 *2 3. 製造所の構造設備の図面 *3 4. GMP 組織図、品質保証体制に関する資料 5. GMP 文書体系 *7 *4 *5 6. 製造工程に関する資料 (1) 代表品目 *1 *6 に係る製造工程のフロー図及び製造方法の詳細に関する資料 (2) 代表品目に係る工程内試験検査項目及び工程管理値に関する資料 (3) 代表品目に係る中間製品並びに製品の規格及び試験方法に関する資料 (4) 原料の受入試験内容及び規格に関する資料 7. バリデーションの実施状況がわかる資料 *8 *9 *10 8. 製造実績あるいは年間予定ロット数 9. 製造所からの出荷に関する手順に関する資料 10. 逸脱管理手順及び実績に関する資料 *12 11. 変更管理手順及び実績に関する資料 *13 12. 生物由来原料基準への対応状況に関する資料 *11 *14 13. (サイトマスターファイル) *に関する説明 *1 平成 20 年 7 月 29 日に品質管理部から示した事務連絡に従い、適合性調査申請 時に添付資料として提出してください。 *2 製造所の立地環境がわかるもの及び製造所全体の施設配置がわかるものを提出し てください。 *3 調査対象となる施設について必要な事項が記載された図面を用意してください。なお、 関係する試験検査施設や動物飼育舎等関連施設についても含めてください。 人・資材等の各種動線、施設内の環境管理の区分、室間差圧の状況をわかりやすく記 載することとし、施設内の環境管理の区分については、空調の系統の区別についても記 載してください。 *4 責任者の会社内の職責及び氏名を記載してください。製造所以外の品質保証部門が 関与する場合は、その関連がわかるように記載してください。 *5 GMP に関係する文書がどのような体系にて管理されているのかがわかるように題名、 文書番号等で整理し、その体系がわかるリスト等で説明したものを用意してください。 *6 当該品目に係る基準書又は手順書の写し(例えば製造所で実際に運用している製造 指図書原本、製造記録、試験記録及び製造・試験検査の手順書の写し等)の該当部分等 を提出してください。 また、再加工、再処理の工程が決められているものは、それらにつ いても記載してください。 *7 製造所の区分が複数ある場合は、区分ごとに 1 品目を代表品目として選定してくださ い。 *8 承認規格と自社規格の区分を明記してください。 *9 原薬等登録原簿(MF)で規格を設定した原料又は承認申請書の「成分及び分量又は 本質」欄に記載した原料について提出してください。 *10 平成 17 年 3 月 30 日付薬食監麻発第 0330001 号、第 3 章第 4 バリデーション基準 の別紙 3-4-2 を参照してください。 *11 製造所からの出荷判定の責任者の規定がされた文書の提出も含めてください。 *12 実績に関する資料とは、申請品目関連で申請の日から過去 2 年以内に行った逸脱 管理に関する記録を提出してください。 *13 実績に関する資料とは、申請品目関連で申請の日から過去 2 年以内に行った変更 管理に関する記録を提出してください。 *14 生物由来原料基準の対象となる原料がない場合には、その旨を記載してください。 ○その他の留意事項 1.上記は標準的なものであり、品目や対象工程により要求資料が変更になる場合があります。 実際には調査員の照会に従ってください。 2.外国語の資料が大部に及ぶ場合、概要を日本語により作成することで差し支えありませ ん。 3.資料について、製造業者等から直接、総合機構に提出することも可能です。 4.過去 2 年以内に機構の調査(書面調査を含む。)を受けており、同様の資料を提示済の場 合は、前回提出以降の変更があったもののみの提出で可能です。その場合、前回提出日を 記載のうえ、提出してください。 5.MRA/MOU 対象国による GMP 適合性証明書(原本に限る。)が添付できる場合は、上記の 資料のうち、2~11 についての提出は不要です。 ここで、MRA とは Mutual Recognition Agreement のことで、対象となる国とは以下の国をさし ます。(ただし、無菌医薬品・バイオ医薬品を除く製剤たる医薬品の工程が対象) ベルギー、デンマーク、ドイツ、ギリシャ、スペイン、フランス、アイルランド、イタリア、ルクセン ブルク、オランダ、オーストリア、ポルトガル、フィンランド、スウェーデン、英国 また、MOU とは Memorandum of Understanding のことで、対象となる国とは以下の国をさしま す。 ドイツ、スウェーデン、スイス、オーストラリア 6.PIC/S に示されている様式等に従ったサイトマスターファイル(概要は日本語訳を添付する こと。)が提出され、2~5、9~11 の記載が含まれる場合についてはこれらの提出は不要です。 ここで、PIC/S とは Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme のことで“製薬業界の監査に関する協力の枠組みを定める協定”のこ とです。詳細は http://www.picscheme.org/を参照してください。 別添2 製造販売業者による GMP 適合性等の確認書 下記のとおり、確認しましたので報告します。 会社名; 報告年月日; 総括製造販売責任者; 許可(認定)番号 許可(認定)年月日 製造所名 製造所所在地 確認実施した者 実施年月日 (他者の場合は 年 月 日~ 年 月 日 会社名記入) 確認対象とした 品目(代表品目 名、製剤/原薬 の別も記載) 医薬品及び医薬部外品の製造管理及び品質管理の基準に関する省令 (通則部分)すべて共通して実施 *コメント欄は自由にお使いください。 項 目 GMP省令 確認の有無 製造部門及び品質部門 第4条 □有 □無 製造管理者 第5条 □有 □無 職員 第6条 □有 □無 製品標準書 第7条 □有 □無 手順書等 第8条 □有 □無 コ メ ン ト 構造設備 第9条 □有 □無 製造管理 第10条 □有 □無 品質管理 第11条 □有 □無 製造所からの出荷の管 第12条 □有 □無 理 バリデーション 第13条 □有 □無 変更の管理 第14条 □有 □無 逸脱の管理 第15条 □有 □無 品質等に関する情報及び品質不 第16条 □有 □無 回収処理 第17条 □有 □無 自己点検 第18条 □有 □無 教育訓練 第19条 □有 □無 文書及び記録の管理 第20条 □有 □無 良等の処理 原薬の製造管理及び品質管理 (該当の有無: 項 ) 目 GMP 省令 確認の有無 品質管理 第21条 □有 □無 文書及び記録の管理 第22条 □有 □無 コ メ ン ト ン ト 無菌医薬品の製造管理及び品質管理 (該当の有無: 項 ) 目 無菌医薬品の製造所の 構造設備 GMP 省令 第23条 確認の有無 □有 □無 コ メ 製造管理 第24条 □有 □無 教育訓練 第25条 □有 □無 生物由来医薬品等の製造管理及び品質管理 (該当の有無: 項 ) 目 GMP 省令 生物由来委託品の製造 確認の有無 第26条 □有 □無 製造管理 第27条 □有 □無 品質管理 第28条 □有 □無 教育訓練 第29条 □有 □無 文書及び記録の 第30条 □有 □無 第31条 □有 □無 コ メ ン ト 所の構造設備 管理 記録の保管の特例 製造販売承認書との齟齬 (該当の有無: 項 目 ) 確認の有無 製造方法* □有 □無 工程管理値* □有 □無 試験規格* □有 □無 試験方法* □有 □無 その他* □有 □無 コ メ ン ト *MFを利用している場合は、MF管理人等が実地で確認したうえで、GQPの取り決め内容との 齟齬について記載すること。
© Copyright 2026 Paperzz