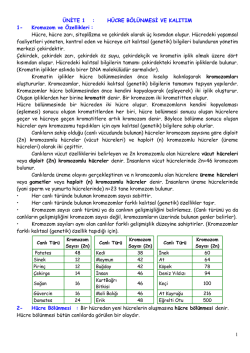
Kanserin Moleküler Temeli ve Jinekolojik Kanserlerde Önemi 19 Dr. Ayfle Ayhan GENET‹K ZEDELENME, KANSERE PRED‹SPOZ‹SYON, A‹LEV‹ KANSERLER Kanserin hücre büyümesi, ölümü, hatta yafllanmas› ve DNA tamiri genleri gibi çeflitli genetik de¤ifliklikler sonucu geliflti¤i düflünülmekte, karsinojenlere maruziyet sonras› ortaya ç›kabildi¤i bilinmektedir. Mutasyonlar kal›tsal olabilece¤i gibi eksojen nedenlerle de olabilir. Genellikle kanser insidans› yaflla birlikte artar, çünkü insanlar yaflland›kça, hücre transformasyonuna kadar ilerleyen çeflitli genetik zedelenmelerle karfl›laflma olas›l›¤› artar. Bir hücrenin tam transformasyonu için onkogen aktivasyonu ve tümör bask›lay›c› gen inaktivasyonu ya da DNA tamir genleri ve apoptoz genlerini de ilgilendiren birçok genetik de¤iflikli¤in oluflmas›n›n flart oldu¤u düflünülmektedir (1). Hastalar s›k olarak ailedeki kanserli akrabalar›n› düflünüp kendi kanser olma ihtimalinin ne kadar oldu¤unu sorar. Bugünkü bilgilere göre her ne kadar toplumdaki kanserlere neden olan genetik de¤iflikliklerin ço¤u sonradan kazan›lm›fl (akkiz) ise de, kanserle ilgili genlerdeki kal›tsal mutasyonlar›n da kanser nedeni oldu¤u gayet iyi bilinmektedir, bu tip sorulara çok dikkatli ve iyi inceleyerek cevap vermek gerekir. Örne¤in akci¤er kanseri ço¤unlukla aç›k ve seçik olarak sigara ile iliflkili iken, akci¤er kanserli hastalar›n sigara içmeyen ana-baba-kardefllerinde akci¤er kanseri mortalitesi (bu kiflilerde pasif içiciligin etkisi yads›namasa da), kontrollerden dört kat fazlad›r. Üstelik kansere neden olan “kal›tsal mutasyonlar” kanser hastalar›n›n %10’undan az›nda bulunur. Buna ra¤men kanserin kal›tsal predispozisyonu, bu hastal›¤›n patogenezini anlamam›za son derece yard›mc› olmufltur. Kansere genetik predispozisyonu üç s›n›fta toparlayabiliriz (2-5): DNA Tamirinin Defekti Nedeni ‹le Oluflan Sendromlar DNA tamir mekanizmas› bozuklu¤u nedeni ile DNA stabilitesi kaybolur, bu grup hastal›klar genellikle otozomal resesif kal›t›m gösterir. Xeroderma pigmentozum ve Ataksi -Telenjiektazi bu gruptad›r. En s›k rastlanan kanser predispozisyonu sendromu olan HNPCC (kolon, ince barsak, endometrium ve over kanserlerine yatk›nl›kla giden Lynch sendromu) otozomal dominant geçifllidir ama bu gruptad›r (5). Ailevi Kanserler Yukar›da bahsedilen kal›tsal sendromlar›n d›fl›nda baz› ailelerde kesin geçifl yolu belirgin olmayan bir kanser yatk›nl›¤› gözlenir. Ailevi kanserleri karakterize eden özellikler erken yaflta görülmeleri, indeks hastan›n iki veya daha fazla birinci derece yak›n›nda ortaya ç›kmas› ve senkron veya metakron, multipl ya da bilateral kanserlerin görülmesidir. Bu gruptaki kanser yatk›nl›¤› muhtemelen çok say›da düflük penetransl› allelin etkisi ile oluflmaktad›r (2,4). Bununla birlikte, önemli olmas› nedeni ile tekrar etmek istedigimiz bir konu, yukar›da tan›mlanan kanserle iliflkili genlerin taranmas›n›n, hangi fertte kanser geliflece¤inin bilinmesi ve buna karfl› önlem al›nmas› aç›s›ndan (ailevi medüller tiroid kanser›nde ret mutasyonlar›n›n saptanmas› d›fl›nda) bugün için tedavi ve prognozda yarar sa¤layamad›¤›d›r, çünkü bu genlerin penetrans› tam olarak anlafl›lm›fl de¤ildir. Akkiz kanserlerde oldu¤u gibi mutasyon tafl›y›c› kiflilerde geliflen ailevi kanserler de ek genetik de¤iflikliklerin birikimi sonucu geliflmektedir. Hangi organda tümör olaca¤›n› kestirmenin mümkün olmamas› yanıs›ra henüz önlem için gelifltirilmifl bir ilaç da yoktur. Otozomal Dominant Geçiflli Ailevi Kanser Sendromlar› Kal›tsal Olmayan Predizpozisyonlar Tek bir otozomal dominant genin kal›t›m› ile tümör geliflen kanser sendromlar›nda mutasyonlar, genelde tümör bask›lay›c› genlerde oluflan nokta mutasyonlar›d›r. Çocukluk ça¤› retinoblastomas› bu grubun en iyi örne¤idir. Kal›tsal kanser sendromlar›nda genelde tümör seçici olarak baz› organ ve dokularda yerleflme e¤ilimi gösterir, inkomplet penetrans ve de¤iflken gen ekspresivitesi olabilir (Tablo 19-1). Ailevi ya da kal›tsal olmayan predizpozisyonlara gelince, söylenebilecek en güzel söz “kansere yakalanmamak için en kesin yol hiç do¤mamakt›r, yaflamak kansere yakalanma riski ile gider” cümlesi olabilir. Baz› çevresel, davran›flsal ve klinik durumlar kanser riskini art›r›r. Örne¤in kronik iltihap ve regenerasyon ile giden durumlar yan›s›ra pernisiöz anemi, solar keratoz, vulva lökoplakisi gibi baz› non-neoplastik durumlar kan- 1 2 J‹NEKOLOJ‹K ONKOLOJ‹ TABLO 19-1 Baz› Kal›tsal Ailevi Kanser Sendromlar› Sendrom ‹smi Gen Kromozom-Yerleflim Predominant Kanser Otozomal Dominant Sendromlar Ailevi meme-over Retinoblastoma Familial polipozis Koli Li-Fraumeni Wilms Tümör MEN2 MEN1 HNPCC (Herediter nonpolipozis kolorektal kanser) Cowden Sendromu Von Hippel Lindau BRCA1 BRCA2 Rb APC p53 WT1 ret 17q21 13q12 1q14 5q21 17q13 11p13 10q11 11q13 /meme-over, kolon, tuba /meme, over, tuba, prostat Retinoblastom, sarkomlar Kolorektal, tiroid Meme, beyin, mide, sarkomlar Böbrek Tiroid, feokromasitom Adrenal, pankreas adac›k hMSH2 hMLH1 hPMS1 hPMS2 PTEN VHL 2p16 3p21 2q31 7p22 10q23 3p25 En çok kolorektal, endometrium, over, hepatobilier, ureteropelvik, beyin, deri hMSH2 ve hMLH1 için ilaveten deri ekleri Hamartoma+endometrium, meme, tiroid vasküler, böbrek-RCC 9q,3p,19q,15p 11q22 15q26 15 farkl› gen Bazal veya skuamöz deri, melanoma Lenfoma, lösemi, karsinom Lenfoma, lösemi, karsinom, çocuk ça¤› AML, MDS Otozomal Resesif, DNA Tamir Defekti ile ‹lgili Sendromlar Xeroderma Pigmentozum Ataksi telenjiektazi Bloom sendromu Fankoni anemisi XP ATM BLM FANCAtoM Ailevi Kanserler (Ailevi s›kl›k var ama kal›tsal predispozisyon henüz bilinmiyor) Meme Over Pankreas ser ile çok yak›n iliflkili olduklar›ndan “prekanseröz” olarak isimlendirilirler. Kazan›lm›fl (akkiz) kanserlerde ise genetik de¤ifliklikler genellikle çevresel etkenlerle olmaktad›r ve bu etkenler viral (örne¤in HPV-serviks kanseri; HBV-hepatosellüler kanser), kimyasal (metilasyon, deaminasyon, DNA hidrolizi s›ras›nda endojen mutasyonlar), fiziksel (ultraviyole ›fl›nlar›) olabilir. Ayr›ca sigara duman›n›n da pek çok kansere neden oldu¤u bilinmektedir. Bununla birlikte kanserlerin ço¤unda direkt bir neden-sonuç iliflkisi henüz ortaya konulamam›flt›r (2). Kanserin Moleküler Temeli Malign tümörler her ne kadar genelde makroskopik, mikroskopik ve genetik materyel bak›m›ndan farkl›l›klar gösterseler de, kendi aralar›nda büyüme ve biyolojik davran›fllar›n› belirleyen baz› -karakteristik- özellikleri paylafl›rlar (2,3,6). ‹ster kal›tsal ister çevresel etkenlerle olsun, temelde hücrenin genetik materyelini ilgilendirdi¤i için, kanser genetik bir hastal›k olarak kabul edilmektedir. Karsinogenezdeki ana mekanizmalar›n özünde, hücredeki “öldürücü olmayan genetik zedelenme” bulunur. Bu DNA zedelenmesi (yani mutasyon) kimyasal-radyasyon-virusler gibi çevresel etkenlerle sonradan olabilece¤i gibi, germ hücreleri ile de kal›t›labilir. Böylece uygun ve yeterli bir genetik zedelenmeye u¤rayan hücreler klonal olarak ço¤al›r (yani tümörler monoklonaldir) tümörü oluflturan klon içerisindeki hücreler otonom hareket eder ve her hücrede bu s›rada instabilite nedeniyle oluflan ilave genetik de¤ifliklikler aras›nda bulunan “h›zl› büyüme, damarlanmay› indükleme, invazyon ya da metastaz yapma, konakç› direnç mekanizmalar›ndan kaçabilme, antineoplastik ilaçlardan etkilenmeme” gibi özellikeri olan hücreler di¤erlerinden daha üstün olup büyüme flans› kazanacak ve onlardan oluflan klonlar sayesinde tümör zaman içerisinde ‘klonlar içinde klonlar’ halinde büyümeye devam edecektir. Bu biyolojik olay, bafllang›çta monoklonal olan tümörün art›k klinik olarak tespit edilebilir hale geldi¤inde heterojen bir hücre popülasyonundan oluflmas›na neden olur (7). Fonksiyonlar› hücre proliferasyonu ile ilgili dört farkl› tip gende de¤ifliklik olur. K›saca, proliferasyounu indükleyenlerde aktivasyon, bask›layanlarda inaktivasyon; hücrenin programl› ölümünü ilgilendiren genler (apoptoz genleri) ve DNA tamir genlerinde bozulma en önemlileridir. Karsinogenez, çok say›da mutasyonun birikimi ile giden, fenotipik düzeyde oldu¤u gibi, genetik bak›mdan da çok basamakl› bir olayd›r (6) (fiekil 19-1). Normal Hücre Ço¤almas› ve Tümör Büyümesinin Biyolojisi Hücrenin genetik materyelinde oluflan de¤ifliklikler esas olarak ‘anormal’ proliferasyona yol açt›¤› ve kanserin esasta bir anormal proliferasyon oldu¤unu düflünerek, ‘normal’ proliferasyon BÖLÜM 19 ❏ Kanserin Moleküler Temeli ve Jinekolojik Kanserlerde Önemi fiEK‹L 19-1 3 Karsinogenezde temel mekanizmalar. ve ilgili genler ile, tümör hücresinin büyüme biyolojisini k›saca hat›rlamak yararl› olacakt›r. Bir hücre popülasyonunda bulunan hücre say›s›n› etkileyen en önemli faktörler proliferasyon h›z› ve ölüm h›z›d›r. Anormal proliferasyonun engellenmesi ise DNA sentezinin ve hücre bölünmesinin engellenmesi ile olmaktad›r. Hücre için ço¤alman›n uygun ya da gerekli oldu¤u durumlarda ise inhibitör mekanizmalar engellenir ve büyüme/proliferasyon sinyalleri a盤a ç›kar. TABLO 19-2 Normal Hücre Proliferasyonu Basamaklar› • Hücrenin proliferasyon için karar vermesi, ilgili ve gerekli molekülleri sentezlemeye haz›rlanmas› • Büyüme faktörlerinin sentezlenmesi ve yeni sentezlenen büyüme faktörünün -kendisine has- büyüme faktörü reseptörüne ba¤lanmas› • Proliferasyon sinyalinin bu reseptörlerden, sinyal iletici moleküller arac›l›¤›yla nükleusa tafl›nmas› • Nükleusta DNA eflleflmesi/ço¤almas› Normal Hücre ve Tümör Hücresi Ço¤almas› Karfl›laflt›rmas› Normal hücre proliferasyonu ancak o hücrenin ve o hücrenin bulundu¤u organ›n uygun koflullar›nda, çevresel uyaranlar›n da uygun oldu¤u koflullarda hücrenin nükleusu taraf›ndan karar verilerek oluflan –kontrollü- bir olayd›r. Basite indirgeyerek özetleyelim (3,8) (Tablo 19-2). Bu basamaklar incelendi¤inde flu sonucu ç›karabiliriz: Baz› hormonlar da dahil omak üzere, hücre d›fl› yerleflimli büyüme faktörleri; transmembran nitelikteki büyüme faktörleri reseptörleri; intrasitoplazmik sinyal iletici moleküller ve nükleer transkripsiyon faktörleri onkogenik aktivasyon sonucu uyar›ld›klar›nda hücre büyümesini indükler. Görüldü¤ü üzere genin hücre içi lokalizasyonu ile fonksiyonu aras›nda bir iliflki yoktur. Yine hücresel lokalizasyona bak›lmaks›z›n proliferasyonu engelleyici genlerde kay›p varsa hücre proliferasyonu engellene- 4 J‹NEKOLOJ‹K ONKOLOJ‹ meyece¤inden hücre anormal proliferasyona yönlenecektir. Hücre siklusunda görevli moleküllerden siklinler ve siklin ba¤›ml› kinazlar›n aktive edici mutasyonlar› onkogen, cdk inhibitörleri ise tümör bask›lay›c› gen fonksiyonunu taklit eder. Hücrenin programl› ölümünü ilgilendiren apoptotik ve antiapoptotik genler yan›s›ra DNA tamiri ile ilgili genlerdeki defektler sonucu da benzer de¤ifliklikler kaç›n›lmazd›r. Di¤er bir deyimle onkogen dedi¤imiz genler hangi yerleflimde ya da hangi fonksiyonla olursa olsun afl›r› uyar›lma sonucu, tümör bask›lay›c› genler ise fiziksel veya fonksiyonel olarak bask›lanma sonucu hücrede anormal proliferasyona neden olan genler olarak tan›mlanabilir. Bu genler asl›nda normal proliferasyonda görev almakla birlikte karsinogenez s›ras›nda oluflan mutasyonlar nedeniyle anormal proliferasyona neden olacak fonksiyon kazanmaktad›rlar: Özetle, onkogenlerin bu aktive edici fonksiyonlar› genellikle nokta mutasyonu, translokasyon veya gen amplifikasyonlar› gibi genetik de¤iflikliklerle; tümör bask›lay›c› genlerin ise delesyon veya nokta mutasyonu gibi fonksiyon kayb›na neden olan genetik de¤iflikliklerle olufltu¤unu söyleyebiliriz (2,3). yüyen tümörlerde dahi- bölünen fraksiyon (büyüme fraksiyonu) %20’ye ancak yaklafl›r. Sonuçta tümör büyüme h›z›, hücre bölünme h›z› ve kayb› aras›ndaki denge ile orant›l›d›r. Kayba neden, replikatif havuzu beslenme yetersizli¤i, farkl›laflma ya da istirahat faz›na dönme gibi çeflitli nedenlerle terkeden hücrelerdir. Tümör hücre kineti¤i çal›flmalar›ndan elde edilen sonuçlar bize h›zl› büyüyen tümörlerde hem ço¤alma hem de apoptoz h›z›n›n çok yüksek oldu¤unu göstermifltir, tabiidir ki tümör büyüdü¤üne göre proliferasyon h›z› apoptoz h›z›ndan büyük olmal›d›r. Tümörün büyüme h›z› genelde farkl›laflmas› ile orant›l›d›r, malign tümörler benign tümörlerden çok daha h›zl› büyürler. Bununla birlikte bir bebe¤in ana karn›nda büyümesinin, en malign tümörden dahi çok daha h›zl› oldu¤unu unutmamak gerekir. Konuya dönersek, hormonal stimulasyon, damarlanma ve bugün için bilemedi¤imiz pek çok faktör de tümör büyümesini etkiler. Ayr›ca antikanser ajanlar›n özellikle -bölünen hücrelere- etki etmesi nedeniyle büyüme fraksiyonunun yüksek olmas› kemoterapi ve radyoterapiye cevapta önem tafl›r (2,9). Tümör Hücresinin Ço¤almas› ‹le ‹lgili Genler Tümör Büyümesinin Biyolojisi, Tümör Hücre Büyüme Kineti¤i Tümör do¤al hikayesini anlamak, tümör büyümesini etkileyen faktörler ile klinik ve tedavi cevaplar›na etkisini ilgilendirdi¤inden, tümör biyolojisinde oldukça önem tafl›r. Bu olay› basamaklara ay›racak olursak, tümör hücresinin büyüme kineti¤i, s›ras›yla hücrenin transformasyonu, ço¤almas› (proliferasyon), damarlanma özellikleri, invazyonu ve metastaz› olarak özetlenebilir. Tümör hücre büyüme kineti¤inden k›saca bahsedildikten sonra transformasyon, hücre ço¤almas› ile ilgili faktörler, invazyon ve metastaz sonraki bölümde detayl› olarak anlat›lacakt›r. Tümör Hücre Büyüme Kineti¤i Tümörler “sonsuz ço¤alan bir dinamo” de¤ildir. Klinik olarak tesbit edildiklerinde hayat sikluslar›n›n önemli bir k›sm›n› tamamlam›fllard›r. Bu durum kanser tedavisinde en önemli sorundur ve kanserin erken tan›s›n›n önemli olmas›n›n esas nedenidir. Tümör büyüme h›z›; tümör hücrelerinin bölünme h›z›, tümör kitlesi içerisinde ço¤alan hücre fraksiyonu ve tümör hücrelerinin ölüm h›z› olmak üzere esasta üç ana faktöre ba¤›ml›d›r. Hücre siklusu tümörlerde anormal hale gelen Rb, p53, siklinler gibi moleküllerce kontrol edilir, bu nedenle sanki tümör hücrelerinin hücre siklusu normal hücrelerden daha k›sa imifl gibi düflünülebilirse de, asl›nda tümör hücrelerinin bölünmesi için geçen zaman normal hücreninkine eflit, hatta daha uzundur. Malign tümörlerde tüm tümör hücrelerinin bölünmekte oldu¤unu düflünmek de bir di¤er yanl›flt›r: her ne kadar tümör büyümesinin çok erken dönemlerinde durum böyle ise de, klinik olarak tesbit edilebilir hale gelen bir tümörde –en h›zl› bü- Bu bölümde s›ras›yla hücrenin kendi kendine yeterli büyüme sinyallerini veren genler, yani “onkogenler”; büyümenin dizginlenmesine duyars›zl›¤a ve yafllanmadan kaç›fla neden olan genler, yani “tümör bask›lay›c› genler”; apoptozu regüle eden, apoptozdan kaç›fla neden olan genler ve -s›n›rs›z ço¤alma potansiyeline neden olan “telomerler” ile “telomeraz” genlerinden bahsedilecektir. Hücrenin Kendi Kendine Yeterli Büyüme Sinyalleri: Onkogenler Biraz önce bahsedilen bilgiler do¤rultusunda onkogenler sadece ve sadece anlat›m kolayl›¤› sa¤lamak için yerleflimlerine göre- hücre d›fl› (büyüme faktörleri), hücre yüzeyi (büyüme faktörü reseptörleri), sitoplazmik (sinyal iletici moleküller) ya da nükleer yerleflimli moleküller olarak s›n›flan›p anlat›lacakt›r. Büyüme Faktörleri ve Büyüme Faktörleri Reseptörleri Büyüme faktörleri çevre hücrelerden veya bizzat tümör hücresinden salg›lanm›fl olabilece¤i gibi endokrin hormonlar da büyüme faktörü olarak görev yapabilirler; mutasyona u¤ray›p tümör geliflimine neden olanlar ONKOGEN, hücrenin proliferasyonundan sorumlu ve mutasyona u¤ramam›fl eflde¤erleri ise PROTOONKOGEN olarak isimlendirilir. Bir tümör hücresinin hem büyüme faktörünü, hem de o büyüme faktörüne has reseptörü salg›lamas› o tümör hücresinin OTOKR‹N büyümesine; ve otokrin büyüme varl›¤› da genelde o tümörün daha agresif biyolojik davran›fl›na neden olmaktad›r (10-18). Büyüme faktöründe mutasyon olmad›¤› durumlarda sinyal iletiminde rol oynayan di¤er genlerdeki mutasyonlar (örne¤in ras) büyüme faktörü genlerinde daha fazla ekspresyona neden olup afl›r› miktarda büyüme faktörü üretimine neden olabilir. Ancak bu olay tek bafl›na neoplastik transformasyon için yeterli de¤ildir. BÖLÜM 19 ❏ Kanserin Moleküler Temeli ve Jinekolojik Kanserlerde Önemi Büyüme faktörü reseptörleri yap›sal olarak genelde her biri kendine has büyüme faktörünü ba¤layan hücre d›fl› parça; hücre zar› boyunca uzanan transmembranöz parça ve tirozin kinaz aktivitesi olan sitoplazmik parçalardan oluflmaktad›r. Tirozin kinaz aktivitesi uyar›lmas›; sinyali, sinyal iletici moleküller arac›l›¤›yla nükleusa iletir. Reseptörler onkogenik hale gelince, büyüme faktörü ba¤lanmas›n› gerektirmeyen kal›c› bir aktivasyon oluflur, yani mutant reseptörler ard› arkas› kesilmeyen ço¤alma sinyalleri verir (2,3,6,11-14,16,17). Çeflitli büyüme faktörleri, kendilerine has reseptörleri ve baz› kanserlerde görülen de¤ifliklikleri Tablo 19-3’te özetlenmifltir. Büyüme faktörü reseptörleri; mutasyonlar, gen rearranjmanlar› ya da afl›r› ekspresyon (over-expression) fleklinde aktive olabilmektedir. Büyüme faktörlerinin normal formlar›n›n afl›r› ekspresyonu, mutasyonlar›ndan daha s›k görülür. Bu afl›r› ekspresyon bazen gendeki amplifikasyona -gen kopyas›ndaki say›ca art›fla- efllik eder. En s›k görülen afl›r› ekspresyon, EGF reseptör ailesi fertlerini (EGFR1=c-erb B1; EGFR2=c-erb B2; EGFR3=c-erb B3; cripto, amfiregülin vb.) ilgilendirmektedir (11-13,16). Örne¤in c-erbB2 geni (Her2/neu geni) meme, over, akci¤er ve mide kanserleri baflta olmak üzere insan adenokarsinomlar›n›n ço¤unda amplifikasyon gösterir ve meme, over, endometrium, serviks ve vulva kanserlerinde kötü prognozun ha- TABLO 19-3 5 bercisidir. Hatta anti c-erbB2 (Herseptin) bugün klinikte kullanilan tedavi seçenekleri aras›na giren moleküllerden biridir. Sinyal ‹letici Moleküller Büyüme faktörünün kendine has reseptörüne ba¤lanmas› ile ortaya ç›kan prolilferasyon sinyali, sitoplazma boyunca nükleusa kompleks bir sinyal iletim mekanizmas›yla ulaflt›r›l›r. Ço¤unlu¤u hücre d›fl›ndan gelen sinyalleri nükleusa iletmede stratejik yerleflim olan hücre zar› iç yapra¤›nda yer alan, biyokimyasal yap›s› heterojen bu grup onkogenlere en iyi örnek, kolorektal neoplaziler baflta olmak üzere insan neoplazilerinde en s›k rastlanan onkogen olma özelli¤ini tafl›yan ras molekülüdür (2,5,6). ‹nsan tümörlerinin %10-20’sinde mutant ras proteini saptanmakla birlikte bu oran kolon, pankreas ve tiroid kanserlerinde çok daha yüksektir. Ras molekülü, G proteinleri ailesinden olup normal proliferasyonda inaktif ras (GDP ba¤lar) aktive olunca (GTP ba¤lar) sinyali nukleusa iletip derhal defosforile olarak inaktif forma geçerken, mutasyon nedeniyle inaktive olamad›¤› durumlar olan malignitelerde tekrarlayan ve s›n›rs›z bir ‘proliferasyon sinyali iletme’ durumunda kalacakt›r. Ras gen mutasyonlar› genellikle genin 12, 13 ve 61 kodonlar›n› ilgilendirmektedir. Ayr›ca ras molekülü, yukar›da bahsedildi¤i üzere EGF ve PDGF gibi büyüme faktörleriyle de iliflki içerisin- Tümörlerde S›k Mutasyon Gösteren Büyüme Faktörleri, Reseptörleri, Sinyal ‹leticiler, Nükleer Proteinler ve Siklus Regülatörleri Büyüme Faktörü Mekanizma De¤ifliklik Gösterdi¤i Tümör Epidermal büyüme faktörü (EGF) Cripto, amfiregülin (CR-1, AR) Heregulin-EGFR2 (c-erbB2) Transforming büyüme faktörü-alfa(TGF-β) Insülin benzeri büyüme faktörleri (IGF-1–2) Makrofaj-koloni stimüle edici faktör (M-CSF) Hepatosit Büyüme Faktörü (HGF) Fibroblast Büyüme Faktörü (FGF-hst-1) afl›r› afl›r› afl›r› afl›r› afl›r› afl›r› afl›r› afl›r› mide,kolorektal mide,kolorektal over, endometrium, meme over, mide over, mesane, kolorektal endometrial, over mide, serviks, over mide, mesane, meme, melanom ekspresyon ekspresyon ekspresyon ekspresyon ekspresyon ekspresyon ekspresyon ekspresyon Büyüme Faktörü Reseptörleri EGFR (erbB1) c-erbB2 HGFR (c-met) M-CSFR (fms) ret amplifikasyon amplifikasyon afl›r› ekspresyon nokta mutasyonu nokta mutasyonu astrositom, osteosarkom, özofagus (skuamöz) meme, akci¤er, serviks, endometrium, over, mide, serviks, over lösemiler, endometrium ca MEN2A, B, FMTC, papiller tiroid Sinyal ileticiler Mekanizma Görüldü¤ü Tümör GTP ba¤lay›c› proteinler (ras) Nonreseptör Tkinaz (abl) nokta mutasyonu translokasyon kolon, pankreas, akci¤er, serviks, endometrium KML, ALL Nükleer Regülatör Protein Mekanizma Görüldü¤ü Tümör myc translokasyon Burkitt, özofagus (skuamöz), serviks Siklus Regülatörü Mekanizma Görüldü¤ü Tümör Siklin D Siklin E CDK4 P27 amplifikasyon overekspresyon amplifikasyon kay›p meme, karaci¤er, over meme melanom,sarkom over (seröz ca) ve birçok tümör 6 J‹NEKOLOJ‹K ONKOLOJ‹ dedir, hücre siklusu regülasyonunda da görev yapar. Ras’a ilaveten, ras sinyal iletim yolunun BRAF ve MAP kinase gibi di¤er molekülleri de kanser hücrelerinde benzer fonksiyonel de¤iflikliklerle sonuçlanan mutasyonlara u¤rayabilir. Pek çok kanserde mutant oldu¤undan, üzerinde çok cal›fl›lmas›na ra¤men henüz klinik kullan›m için umut verici bir anti-ras moleküler tedavisi mevcut de¤ildir(27). Reseptör Olmayan Tirozin Kinaz Molekülleri Reseptör tirozin kinazlar gibi reseptör olmayan tirozin kinazlar da kromozom rearranjman ve translokasyonlar› ile füzyon geni oluflturarak daimi enzim aktivitesine neden olur, kronik myeloid lösemide görülen ve Philadelphia kromozomunun parças› olan abl geni en güzel örnektir: bcr-abl füzyon geninin tirozin kinaz aktivitesini önlemek için tasarlanan imatinib mesilat ise özellikle kronik myeloid lösemilerin tedavisinde kullan›lan ve ça¤ de¤ifltiren ilaçlardan biridir. Nükleer Transkripsiyon Faktörleri Sinyal iletiminde hedef nükleusta DNA replikasyonu oldu¤una göre, esas görevi DNA replikasyonu ve hücre bölünmesi olan ‘nükleus yerleflimli moleküller’de karsinogenezde son derece önemlidir. myc, jun, fos, rel gibi transkripsiyon faktörü olup siklin genleri gibi büyümeyi indükleyen genleri regüle eden bu genler, hücrenin mitotik döngü içerisinde problemsiz ilerlemesini sa¤layan moleküllere en iyi örneklerdir. Hücrelerin büyüme faktörleriyle iletiflimi sonras› nükleer transkripsiyon faktörleri çok k›sa sürede artmakta ve uyar›ld›klar›nda, ürünleri özel DNA regülatör bölgelerine ba¤lanarak DNA replikasyonu ve hücre bölünmesine neden olmaktad›r. myc geni amplifikasyon ya da translokasyon fleklinde mutasyona u¤rar. myc molekülü sentezlenir sentezlenmez nükleusa tafl›n›r, bu s›rada max molekülü ile heterodimer oluflturur. myc-max heterodimerlerinin potent bir transkripsiyon aktivatörü olmalar› nedeniyle, mycmax ba¤lanmas›n› engelleyen mutasyonlar onkogenik aktiviteyi azaltmaktad›r. Üstelik, mad isimli di¤er bir regülatör protein de max’a ba¤lanabilmekte ve max-mad heterodimerleri transkripsiyonu bask›lmaktad›r. Di¤er bir deyimle, transkripsiyon aktivasyonu sadece c-myc’e de¤il, max ve mad proteinlerinin varl›¤› ve miktar›na da ba¤l›d›r. myc molekülü ile ilgili çok önemli di¤er bir konu da, sadece hücre büyümesini konrol eden bir molekül olmay›p apoptoz ile de iliflkili olmas›d›r. Yaflam sinyallerinin (büyüme faktörlerinin ve hormonlar›n) yetersiz kald›¤› ya da olmad›¤› durumlarda myc aktive olup hücreyi apoptoz ile ölüme de götürmektedir. Bunlar d›fl›nda myc geni taraf›ndan modüle edilen çok say›daki hücre fonksiyonu aras›nda hücreler aras› adhezyon azalmas›, motilite art›m›, telomeraz aktivitesi art›m›, protein sentezi art›m› gibi hücrenin h›zla ço¤almas›na katk›da bulunan metabolik de¤ifliklikler de bulunmaktad›r. K›saca, onkojenik myc, persistan ekspresyon oluflturup neoplastik transformasyona efllik etmekte, apoptozla hücre ölmüne giden yola¤› da açamamaktad›r. c-myc amplifikasyonu meme, kolon, akci¤er ve serviks kanserinde görülmektedir (2,3,6). Hücre Siklusu Regülatörleri, Siklin ve Siklin Ba¤›ml› Kinazlar Hücre siklusu regülasyonunu, hücre döngüsünün tüm fazlar›nda ortamda bulunmalar›, yap›m ve y›k›mlar›n›n döngüsel olmas› nedenleriyle siklin ismi verilen moleküller ile siklusun sadece baz› dönemlerinde ortaya ç›karak siklinlere ba¤lanan ve onlar› y›kan siklin ba¤›ml› kinaz (CDK) molekülleri sa¤lar. Ço¤alma sinyalleri, siklin+siklin ba¤›ml› kinaz kompleksi oluflunca Rb gen ürününü fosforile ederek, ortamda inhibitör moleküllerin bulunmad›¤› durumlarda hücre proliferasyonuna neden olurken, hücre proliferasyonunu engelleyen bir durum varl›¤›nda cdk inhibitörlerinin (CDKI) ifle kar›flmas›yla hücre proliferasyonunu engelleyeceklerdir (fiekil 19.1). Di¤er bir deyimle siklinler CDK art›m›na neden olurken; CDKI, CDK azalmas›na neden olarak hücre döngüsünde negatif kontrol oluflturur. CDKI ailesinin bir grup üyesi (p21, p27, p57-yani CIP/WAF ailesi) tüm CDK’lar› genel olarak inhibe ederken; p15, p16, p18 ve p19 seçici olarak siklinD/CDK4 ve siklinD/CDK6 kompslekslerini inhibe eder, ikinci grup INK4 proteinleri olarak adland›r›l›r. Bu k›sa bilgilerin ›fl›¤›nda siklin ve CDK aktivitesi regülasyonunu bozan mutasyonlar›n hücre proliferasyonuna neden olacaklar› aç›kt›r. SiklinD geni afl›r› ekspresyonu meme, özofagus ve karaci¤er baflta olmak ve over kanseri de dahil olmak üzere pek çok kanserde, cdk4 amplifikasyonu ise melanom, sarkomlar ve glioblastomda s›k görülmektedir. CDKI molekülleri genellikle bir çok insan malign tümöründe mutantt›r ya da susturulmufltur. Ayr›ca siklinD art›fl› ile birlikte giden p27 kayb›, meme ve over kanserlerinde gerek sa¤kal›m gerekse kemoterapi cevab›nda ba¤›ms›z bir prognostikatördür. p16INK4A ise serviks non-neoplastik lezyonlar› ile intraepitelyal neoplazilerin ay›r›m› yan›s›ra intraepitelyal neoplazilerin karsinoma progresyonunu öngörmede önem tafl›r. Son y›llarda p27 molekülünün tümör hücre migrasyonunda da önemli rolü oldu¤unu bildirilmektedir (19). Anlafl›lan bu moleküller zaman içerisinde daha detayl› incelendikçe, iliflkili olduklar› di¤er molekülleri ve bugün bilmedi¤imiz fonksiyonlar›n› çok daha iyi anlayabilece¤iz. Hücre döngüsü regülasyonunu takiben k›saca “CHECKPOINT” ad› verilen hücre döngüsü polisleri olan kontrol noktalar›na de¤inelim. Biri G1/S di¤eri ise G2/M geçisinde olmak üzere iki adet checkpoint vard›r. S faz›, döngünün geri dönüflü olmayan noktas›d›r: Hücre ço¤alma karar› verince G1/S noktas›nda DNA zedelenmesi olup olmad›¤› kontrol edilir, e¤er zedelenme varsa döngü beklemeye al›n›p DNA tamir mekanizmalar› harekete geçirilir, zedelenmenin tamir edilemedi¤i durumlarda ise hücrenin programl› ölümü yani apoptoz mekanizmalar› devreye girer. E¤er kromatidler hala ayr›lmam›flsa DNA zedelenmesi, replikasyondan sonra da tamir edilebilir. G2/M noktas› ise DNA replikasyonunun tamamlanm›fl oldu¤unu monitorize eder ve hücrenin emniyetli bir flekilde mitoza girip kromatidlerin ayr›lmas›na f›rsat verir. Radyasyona maruz kalan hücrelerde bu nokta özellikle önem tafl›r: Hücre döngüsünün checkpoint noktalar› düzgün fonksiyon görebilmek için DNA zedelenme alg›lay›c› (RAD ailesi, Ataksi telenjiektazi gen ailesi) ve bu sinyali iletici moleküller (CHK kinaz ailesi) yan›s›ra p53, p21 gibi efektör moleküllere ihtiyaç gösterir. Bahsedilen hücre döngüsü checkpoint komponentlerindeki bozukluklar, kanser hücrelerindeki genetik instabilitenin en önemli nedenidir (2,3,6,20). BÖLÜM 19 ❏ Kanserin Moleküler Temeli ve Jinekolojik Kanserlerde Önemi Büyümenin Dizginlenmesine Duyars›zl›k, Yafllanmadan Kaç›fl: Tümör Bask›lay›c› Genler Onkogenler aktive olduklar›nda hücre proliferasyonunu uyar›rlarken, tümör bask›lay›c› genler hücre proliferasyonunu engellerler, bir anlamda fren olufltururlar. Asl›nda ana fizyolojik fonksiyonlar› tümör oluflumunu engellemek de¤il, normal hücre proliferasyonunu durdurmak oldu¤undan ‘Tümör bask›lay›c› gen’ tabiri yanl›flt›r denebilir. Gene de, bu terim, söz konusu moleküllerin ilk kez tümörlerde bulunup tan›mlanmalar› ve kay›plar›n›n tümöre yol açmas› nedeniyle hala aynen kullan›lmaktad›r. Tümör bask›lay›c› genler aras›nda ilk kez retinoblastom geni tan›mlanm›fl, Knudson’un –iki darbe hipotezi- ile bir tümör bask›lay›c› genin tamamen fonksiyonunu kaybetmesi için her iki allelinin de inaktive olmas› gerekti¤i öngörülmüfltü. Bugün için bu kural›n baz› tümör bask›lay›c› genler için flart olmad›¤›n› da biliyoruz (NF-1 ve APC gibi). Rb geni d›fl›nda üzerinde en çok çal›fl›lan tümör bask›lay›c› genler aras›nda p53 geni, adenomatozis polipozis koli (APC) geni, Nörofibromatozis 1 ve 2 genleri (NF-1 ve NF-2), meme-over kanseri 1 ve 2 (BRCA-1, BRCA-2) say›labilir (2,3,6,20) (Tablo 19-4). Büyümenin dizginlenmesinde bozukluk, karsinogenezdeki temel de¤iflikliklerdendir. Asl›nda, normal flartlarda bir hücrede onkogen ekspresyonu, o hücreyi tümör bask›lay›c› genler sayesinde ya kal›c› bir hücre döngüsü arrestine götürür, ya da ço¤alma potansiyeli olmayan iyi farkl›laflm›fl bir hücreye dönüfltürür. Biyokimyasal ve fonksiyonel olarak tümör bask›lay›c› genlerin ifllevleri henüz onkogenlerinki kadar detayl› olarak anlafl›labilmifl de¤ildir, ama zaman içerisinde farkl› görevleri ve di¤er moleküllerle iflbirlikleri konusunda bilgimiz artmaktad›r. Yine yerleflimlerine göre inceleyelim. Hücre Yüzey Reseptörleri Hücre yüzeyinde yer al›p büyümeyi dizginleyen genler, büyümeyi inhibe eden faktörlerin reseptörleri (örn. TGF-βR) veya kadherinler gibi hücreleraras› iliflkiyi (adhezyon) sa¤layan mo- leküller olabilir. TGF-β’n›n reseptörlerine ba¤lanmas› ile CDKI uyar›l›r ve büyümeyi engelleyici gen ürünleri sentezlenir. TGFβ kuvvetli bir proliferasyon inhibitörüdür, ancak sadece TGFβ molekülünün “kendisi” de¤il, bu yolak boyunca bulunan di¤er moleküller [TGF-β reseptörleri, R-SMAD molekülleri ile efektör CDKI (p21 ve p15)] ya da TGF-β sinyallerinin represe ederek etki gösterdigi siklin ve CDK molekülleri mutasyonlar› da benzer sonuca götüren nedenler olacakt›r. Kolon, mide ve endometrium kanserlerinde TGF-β tipII reseptör mutasyonlar› izlenirken, pankreas kanserinde SMAD4 inaktivasyonu vard›r; pankreas kanserlerinin tamam›nda ve kolon kanserlerinin %80’inden ço¤unda mutlaka TGF-β yolunun en az›ndan bir komponenti mutantt›r. Kadherinler de epitelyal hücre adhezyonunda görev alan moleküllerdir ve kay›plar› hücrenin lokal invazyon ve metastaz›n› kolaylaflt›r›r. Özofagus, kolon, meme ve over kanserinde E-kadherin ekspresyon kayb› s›kt›r. Ayr›ca E-kadherin kayb› ailevi mide kanseri yatk›nl›¤›na da neden olur (2,5,6,20-24). Sinyal ‹letimi ‹le ‹lgili Genler NF-1 ve APC/β-katenin bu kategoride yer al›r. Ailevi olarak söz konusu genlerin birer alleli eksik do¤an kifliler, tümör bask›lay›c› gen kavram›na bir anlamda ayk›r› düflecek flekilde fenotipik olarak normal fertlerden farkl›d›rlar: NF-1 yoklu¤unda nörofibromlar APC yoklu¤unda ise kolonda yüzlerce hatta binlerce adenomatöz polip gelifltirirler; bu benign tümörlerde ayn› genlerin ikinci allellerini de etkileyen ilave mutasyonlar ile bu benign neoplazmlar malign neoplaziye dönüflür. APC geni etkisini, sitoplazmadan nükleusa geçerek hücre bölünmesini sa¤layan myc, siklinD gibi ço¤almay› uyaran genlerin sentezlenmesine neden olan “β-katenin” molekülüne ba¤lan›p onu parçalamakla yapar. APC inaktivasyonu β-katenin düzeylerini art›r›p hücrenin ço¤almas›na neden olmaktad›r. Ayr›ca APC proteini hücre adhezyonu ve migrasyonundan da sorumludur. Kromozom 5q yerleflimli bu gen sadece ailevi polipozis sendromlar›nda de¤il, sporadik kolorektal adenom ve karsinomla- TABLO 19-4 Baz› Tümör Bask›lay›c› Genler, Fonksiyonlar› ve Kay›plar›n›n S›k Görüldü¤ü Tümör Tipleri Yerleflim Gen Hücre yüzeyi TGF-βReseptör E-kadherin Sitoplazmik NF-1 NF-2 APC/β-catenin PTEN SMAD2 ve 4 Rb Nükleus p53 WT-1 p16/INK4A BRCA-1 BRCA-2 FHIT Di¤er 7 Fonksiyon Somatik mutasyonundaki tümör Kal›tsal mutasyonundaki tümör Büyüme dizginleyici Hücre adhezyonu ras sinyal dizginleyici ? sinyal iletim dizginleyici PI3K sinyal iletimi TGF-βsinyal iletimi siklus regülatörü siklus/apoptoz regülatörü nükleer transkripsiyon CDKI DNA tamir DNA tamir Fragil Histidin Triad Kolon Mide, meme, kolon Schwannoma Schwannom ve meningiom Mide,kolon,özofagus, pankreas Endometrial ve prostat kolon ve pankreas retinoblastom, meme, kolon, over pekçok kanser Wilms tm pankreas, meme, özofagus, over özofagus, mide ? Ailevi mide NF-1 & sarkom NF-2, meningiom FAP/kolon Cowden sendromu ? Retinoblastom Li-Fraumeni send. Wilms tm malign melanom /meme ve over / meme, mide ? 8 J‹NEKOLOJ‹K ONKOLOJ‹ r›n oluflumunda ve kolorektal karsinogenezin erken döneminde de rol oynar (1,2,5,6,21). NF-1geni ürünü olan nörofibromin, ras proteininin sinyal iletimini regüle eder. K›saca, normal proliferasyonda GDP ba¤layan inaktif ras aktive olunca GTP ba¤lamakta ve sinyali nükleusa iletir iletmez derhal defosforile olarak yeniden inaktif forma geçmektedir. Bu basamakta nörofibromin GTPaz aktive eden bir protein olarak aktive ras molekülünün inaktif hale geçmesinde önemli rol oynar, yani; NF-1 tümör bask›lay›c› gen ürünü yoklu¤unda ras molekülü aktif halde kalacak ve anormal proliferasyona yol açacakt›r (2,3). Yine NF2 genindeki germline mutasyonlar› Nörofibromatozis Tip2 ye neden olur. NF2 ürünü merlin (ya da nörofibromin2) eritrosit membran proteini ile benzerlik gösterir. Her ne kadar merlin bozuklu¤u ile karsinogenez iliflkisi tam olarak bilinmiyor ise de merlini olmayan (ya da bozuk olan) hücrelerde hücreleraras› kontakt molekülleri bozuktur ve hücreler, bu yoldaki sinyallerle ulaflan büyüme bask›lay›c› sinyallere de duyars›zd›r(3). Transkripsiyon/Hücre Siklusu Regülatörü Nükleer Tümör Bask›lay›c›lar Rb, p53, BRCA-1 ve 2, Wilms tümör bask›lay›c› geni gibi önemli tümör bask›lay›c› gen ürünleri nükleer yerleflimlidir. ‹lk keflfedilen, bu nedenle üzerinde en çok çal›fl›lan tümör bask›lay›c› gen olan Rb bir nükleer fosfoproteindir. Her hücre tipinde mutlaka eksprese olur. Aktif hali az fosfor tafl›yan yap›dad›r ve E2F ailesi transkripsiyon faktörlerini ba¤layarak hücre bölünmesini engeller. Fosforu siklin ve CDK komplekslerinden alarak hiperfosforile olmas› ile Rb inaktive olmakta ve E2F faktörlerini ba¤layamamas›; E2F transkripsiyon faktörünün a盤a ç›kmas›na ve hücrenin G1 faz›ndan S faz›na geçifle imkan vermektedir. Ayr›ca normal-hipofosforile Rb, hem histonmetiltransferaz ve histon deasetilaz ve gibi E2F ba¤›ml› kromatin biçimleyen proteinlerle de iliflkide oldu¤undan, kromatini, transkripsiyon faktörlerine yan›ts›z kalacak flekilde biçimleyerek, hem de hücre bölünmesini engelleyen CDKI proteinlerinden p27 stabilizasyonunun kontrolunda yer alarak hücre döngüsünü durdurmada önemli etki yapar. Hücre bir kere S faz›na girerse büyüme faktör stimülasyonu olmasa da bölünmeye devam eder, M faz›nda ise Rb molekülünden fosfor al›narak tekrar defosforile/az fosforile hale getirilmektedir. Ailevi Rb gen delesyonlar›nda erken yaflta, bilateral ve multipl retinoblastomlar, daha az da osteosarkomlar oluflmaktad›r. Somatik Rb mutasyonlar› ise meme, mesane kanserleri, glioblastom ve akci¤er küçük hücreli kanserlerinde bildirilmifltir. Her hücrede Rb bulunmas› ve hücre siklusundaki önemli görevi göz önüne al›nd›¤›nda insan kanserlerinde inaktive edici Rb gen mutasyonlar›n›n neden daha fazla olmad›¤› düflünülebilir, ancak bu mutasyonlara çok benzeyecek sekilde RB protein kayb›n› taklideden di¤er gen bozukluklar› varl›¤›nda Rb kayb›n›n flart olmad›¤› gayet aç›kt›r: asl›nda ya siklinD/CDK4 gibi RB fosforilasyonunu kolaylaflt›rarak veya CDKI inaktivasyonu ile siklin/CDK aktivitesini art›rarak etki eden mutasyonlar ya da Rb mutasyonlar› insan kanserlerinin hemen tümünde mevcuttur (2,20,23). ‹nsan kanserlerinde en s›k de¤iflikli¤e u¤rayan tümör bask›lay›c› gen olan p53 geni 17p13.1 bölgesinde yer al›r. p53 nokta mutasyonlar›, zaman zaman di¤er allelde kay›plarla ile birlikte giderken, zaman zaman da tek alleli ilgilendirse bile oluflturdu¤u baz› nokta mutasyonlar›n›n sonucu üç boyutlu (konformasyon) de¤ifliklikleri ile normal alleldeki fonksiyon kayb›na da neden olacak flekilde inaktive eder. p53 mutasyonlar› kolon, meme, akci¤er, over gibi organlar›n karsinomlar›nda %50 ve daha fazla görülür, ayr›ca sarkom, lösemi ve lenfomalar, sinir sistemi tümörlerinde de s›k olarak görülmektedir(25). Ailevi defektler Li-Fraumeni sendromu olarak bilinen, erken yaflta ve multipl ailesel kanserlere neden olur. p53 mutasyonlar›n›n insan tümörlerinin pek ço¤unda bulunmas›, bu genin ürünü olan proteinin (TP53) kanser oluflumunda önemli bir koruyucu görevi yapt›¤›n› göstermektedir (26). p53’ün birbiriyle çok yak›ndan iliflkili üç görevi hücre siklusunu geçici olarak durdurmak, hücre yafllanmas› da diyebilecegimiz kal›c› olarak durdurmak ya da programl› hücre ölümü yani apoptozu tetiklemek; sonuçta fonksiyonu, genetik olarak hasar görmüfl hücrelerin ço¤almas›n› engellemektir. TP53, p53 geni ürünü protein olup nükleusta yer al›r ve hücre proliferasyonu ile ilgili, di¤er pek çok geni kontrol eder. Rb gen ürününden farkl› olarak her hücre proliferasyonunda görev almaz, sadece acil durumlarda, genetik zedelenmelerin varl›¤›nda (örne¤in UV, radyasyon ya da mutajenik kimyasallar gibi nedenlerle DNA hasar› oldu¤unda) ya da hipokside görev bafl›na geçer. Hasarda p53 proteni hücrede birikir ve ilgili hücreyi hücre siklusunun G1 faz›nda durdurur; hasar onar›lana kadar da etkin kal›r. Bununla birlikte DNA hasar› onar›lmam›fl ise p53 molekülü, (bir k›sm› p21 cdki molekülü arac›l›¤› ile) iliflkide oldu¤u apoptoz genlerinden bax’ ›n uyar›lmas›na ve DNA’s› hasarl› hücrenin apoptoz ile ölümüne neden olur (2-6,25-30) (fiekil 19-2). p53’ün son y›llarda keflfedilen yeni bir fonksiyonu ise baz› proliferasyon öncüsü ya da anti-apoptotik genlerdeki represyondur, bu etkiyi mikroRNA(miRNA)’lar üzerinden yapar: p53, mir34 ailesinin yap›m›n› aktive eder, mir34 ailesi ürünleri ise siklinler gibi proliferasyon öncüsü ya da bcl2 gibi antiapoptotik genlerin mRNAs›nda ilgili bölgelere ba¤lanarak yap›mlar›n› engeller. mir34 microRNAlar hem p53 regülasyonu için gerekli, hem de p53’ün bir çok fonksiyonunu tekrarlayan moleküller oldu¤undan p53 regülasyonundaki önemleri büyüktür (31). K›sacas› p53, genom bütünlü¤ü ve sa¤laml›¤›n›n sigortas›d›r. Genom bütünlü¤ü yukar›da da bahsedildi¤i gibi indirekt olarak sa¤lan›r: DNA hasar› varl›¤› ve onar›m›n yeterlili¤i konular›ndak bilgileri ATM/ATR (Ataksitelenjiektazi genleri); DNA zedelenmesine temel cevap olarak G1 faz› sonunda görülen hücre siklusu durmas› ile DNA tamiri p21 (CDKI) ve GADD45 (Growth Arrest DNA Damage 45); hücre yafllanmas› ile eflde¤er kal›c› hücre siklusu durmas› CDKI ve E2F hedefi genler, DNA zedelenmesinin ortadan kald›r›lamad›¤› durumlarda gereken apoptoz ise BAX, BBC3 gibi genler ile etkileflime girerek baflar›l›r. E¤er p53 allellerinden biri kal›tsal veya kazan›lm›fl olarak kaybolmufl, di¤eri de mutant ise p53 hücrede birikse de anormal protein fonksiyon görmeyece¤inden hücre proliferasyonu- BÖLÜM 19 ❏ Kanserin Moleküler Temeli ve Jinekolojik Kanserlerde Önemi fiEK‹L 19-2 9 (a,b). Normal (vahfli tip) ve anormal (mutant) p53 muhtemel fonksiyon mekanizmalar› na engel olamayacak ve DNA hasarl› bir flekilde hücre proliferasyona devam edecektir. p53 molekülü inaktivasyonu için mutasyon olmas› flart de¤ildir. Adenovirüs E1B proteini p53 molekülüne ba¤lanarak onun aktive etti¤i transkripsiyonu engeller, HPV E6 proteini de p53 protein molekülünü y›kar ve hasar karfl›s›nda proliferasyon engelleyici fonksiyonunu ortadan kald›r›r. Nitekim bahsetti¤imiz etki ayn› virüsün Rb proteinine ba¤lanarak inaktivasyonuna yol açan E7 molekülü üzerindeki etkisi ile birlikte serviks karsinogenezinde son derece önemli bir basama¤› oluflturmaktad›r. K›sacas› p53 molekülü ya mutasyonlarla inaktive oldu¤unda, ya da mutant olmamas›na ra¤men viral partiküller taraf›ndan ba¤lanarak parçaland›¤›nda fonksiyonunu yitirir. Bunlara ilaveten mutant olmad›¤› halde p53 fonksiyon kayb›na neden olan di¤er bir olay MDM2 (murine double minute, insan eflde¤eri hdm) proteinine ba¤lanmas›d›r (32). Bu tip inaktivasyonlarda MDM antagonistleri ile p53 fonksiyonunun geri döndürülmesi bilim adamlar›n›n yo¤un çal›flma konular›ndan birisidir. p53 ailesinin di¤er fertleri olan p63 ve p73’ün keflfi sayesinde p53 kollaboratörleri ile ilgili bilgiler genifllemifl olmakla birlikte kendi aralar›nda belirgin bir “cross-talk” yapan bu kompleks a¤›n oyuncular› hakk›ndaki bilgiler henüz oldukça s›n›rl›d›r. p63 çok katl› yass› epitel ve p73 özellikle kemoterapi sonras› ortaya ç›kt›klar›ndan etkileri daha “doku-spesifik” gibi görünmektedir. Meme kanserinden edindi¤imiz bilgilere göre, her üç molekülün de baz›lar› transkripsiyon aktivatörü di¤erle- ri ise dominant negatif etki yapan ve kendi aralar›nda dengeli ve etkileflen bir koro oluflturan de¤iflik izoformlar› vard›r (2,33). 17q 12-21 bölgesinde BRCA1 geni ve 13q 12-13 bölgesinde de BRCA2 geni vard›r ve isimlerinden de anlafl›laca¤› gibi meme ve over kanseri dahil pekçok kanserin oluflumunda önem tafl›r. BRCA1 gen mutasyonu tafl›yan bir fertte hayat boyunca meme kanserine yakalanma flans› %60 olup; over, prostat ve kolon kanserine yakalanma riski de yüksektir. BRCA2 mutasyonu tafl›yan kiflilerde ise erkek meme kanseri, over kanseri, pankeras ve larinks kanserine yakalanma flans› yüksektir. Bununla birlikte, meme kanserlerinin sadece %5-10 kadar› ailevidir ve BRCA1 ve 2 genleri ailevi olan kanserlerin tamam›ndan de¤il %80 kadar›ndan sorumludur. Buna karfl›l›k sporadik meme kanserlerinde BRCA1 ve 2 gen kayb› gözlenmez (34-36). BRCA1 ve 2 gen fonksiyonlar› da henüz tam olarak anlafl›lm›fl de¤ildir. Her iki gen ürünü de nükleusta yer almakta ve transkripsiyonda regülatör görev ald›klar› düflünülmektedir. Bu genlerin ürünü olan proteinlerin Rad 51 isimli çift sarmal DNA’ n›n radyasyon sonras› tamirinin regülasyonunda görev alan bir genle iliflkili olmas›, bir anlamda DNA onar›m›ndan da sorumlu olduklar›n› düflündürmektedir. Di¤er bir deyimle bu genler hücre büyüme ve proliferasyonundan direk olarak sorumlu olmasa da mutasyonlar› DNA replikasyonunda hatalara neden olaca¤›ndan hücre büyüme ve proliferasyonundan direk olarak sorumlu olan genlerde defektlere neden olur. Son yay›n- 10 J‹NEKOLOJ‹K ONKOLOJ‹ lar, BRCA2 gen ürününün Fankoni anemisinin bir tipinden sorumlu olan FANCD1 oldu¤unu göstermifltir. Ayr›ca di¤er Fankoni ürünleri ve ataksi telenjiektazi (ATM) gen ürünleri de BRCA1 proteini ile iliflkiye girmektedir. Bu nedenle konjenital anomaliler, progresif kemik ili¤i yetmezli¤i ve kansere yakalanma flans›n›n artt›¤› bir sendrom olan Fankoni anemisinden sorumlu di¤er genlerin meme kanseri ile iliflkisinin araflt›r›lmas› son günlerde önem kazanm›flt›r (37,38). Di¤er Tümör Bask›lay›c› Genler PTEN, FHIT ve VHL k›saca özetlenecek olursa (2): Kromozom 10 uzun kolunda yer alan PTEN (Phosphatase and TENsin Homolog) geni endometrium, meme, prostat kanseri ve glioblastomda kay›p göstermektedir. Hem protein hem de lipid fosfatazd›r. Etkisini PI3K/AKT yolu üzerinden yapar. Bu yol normalde büyüme faktörlerinin reseptör tirozin kinazlara ba¤lanmas› sonras› RAS/JAK/STAT yolu ile birlikte uyar›l›r: PI3K, PDK yi aktive eder, o da fonksiyonlar› çok önemli olan serin/threonin kinaz AKT yolunu uyar›r: AKT, p53’te ve apoptozda bahsetti¤imiz BAD ve MDM2 gibi çok say›da substrat› fosforile etmesinin yan›s›ra hücrenin yaflam›n› uzatmakta, ayr›ca tüberoz skleroz tümör bask›lay›c› gen kompleksini (TS1/TS2) inaktive etmektedir. Her ne kadar kazan›lm›fl PTEN fonksiyon kayb› çeflitli kanserlerde en çok görülen PI3K/AKT sinyal yolu aktivatörü ise de, PI3K dahil bu yolda bulunan çeflitli elemanlardaki mutasyonlar› topluca düflünürsek kanserlerde en s›k mutasyon gösteren yol oldu¤u fikrine ulaflaca¤›m›zdan, bu yol ve bu yolu hedefleyen moleküler ilaçlara son y›llardaki artan ra¤betin nedenini kolayca anlayabiliriz (39,40). FHIT: 3p14 yerleflimli bu tümör bask›lay›c› gen frajil bir kromozom bölgesidir (Fragile HIstidine Triad) ve daha ziyade ekzojen karsinojenler taraf›ndan delesyon sonucu inaktive oldu¤u düflünülmektedir. FHIT geninin kodlad›¤› protein hidrolaz aktivitesi olan bir oligofosfatt›r (ApnA) ve hücre diferansiyasyon ve apoptozunda fonksiyonu olan hücre içi sinyal ileticidir. FHIT gen ekspresyonu kayb›n›n meme, mesane, serviks, pankreas ve Barett zemininde geliflen özofagus adenokanserlerinde tümörün ilerlemesi ile iliflkili oldu¤u bildirilmifltir (2,3,41). Von Hippel Lindau (VHL): Üçüncü kromozomun k›sa kolunda yer alan bu gen ailevi renal kanserler, feokromasitoma ve ve sanral sinir sistemi hemanjioblastomlar›, retina anjiomlar› ve renal kistler ile karakterlidir, sporadik renal hücreli kanserlerle de birlikte olabilir. Apoptozu Regüle Eden Genler-Apoptozdan Kaç›fl Neoplastik hücrelerin ço¤alabilmesi için sadece büyümeyi indükleyen genlerin aktivasyonu ya da büyümeyi dizginleyen genlerin inaktivasyonu yeterli olmaz, ayn› zamanda programl› hücre ölümünü regüle eden genlerde de bozukluk olmas› yani kanserli hücrenin ço¤almak için apoptoz engelini de aflmas› flartt›r. Normal flartlar alt›nda genomu zedelenen hücre, programl› olarak ölmeye sürüklenerek mutant hücrelerin ço¤almas› engellenmektedir. Apoptoz regülatörü genler (ço¤u ‘’b’’ harfi ile bafllayan üç harfli genler) mitokondriden sal›nan ve sitokrom C arac›l›¤› ile apoptozu regüle eden gen gurubudur; apoptozu indükleyen (apoptotik: ölüm agonistleri) ya da engelleyen (antiapoptotik: ölüm antagonistleri) genlerdir. Homodimer ya da kompetitif heterodimerler oluflturan bu iki gen ailesi aras›nda antagonistlerin agonistlere oran›, hücrenin apoptotik bir stimulusa nas›l cevap verece¤ini tayin eder. Apoptotik gen ailesi üyeleri aras›nda bax, bak, bcl-XS; apoptozu engelleyen antiapototik gen ailesi üyeleri bcl2, bcl-XL yan›s›ra bad, bid gibi apoptotik ve antiapoptotik proteinler aras›ndaki dengeyi regüle eden genler de bulunur. Bir tümör bask›lay›c› olmakla birlikte ayn› zamanda apoptoz regülatörü olan p53’ün etkisi, düzeltilemeyen DNA hasar› varl›¤›nda hücre ço¤almas›na engel olmak ve hasarl› hücreyi ortadan kald›rmak için, apoptotik bax genini indüklemesinden kaynaklanmaktad›r. Yine nükleer transkripsiyon faktörü onkogenler aras›nda say›l›rken apoptozla iliflkisinden bahsedilen myc geni ise, ancak ortamda yeterli büyüme faktörü varsa hücreyi proliferasyona götürebilmekte, yoksa proliferasyon engellenmekle kalmay›p in vitro ortamda hücreler apoptoza sürüklenmektedir. Bununla birlikte apoptoz regülasyonu her zaman mitokondrial proteinler arac›l›¤›yla olmaz. fas onkogeninin ligand›na ya da Tümör Nekroze edici Faktör’ün (TNF) kendi reseptörüne ba¤lanmas›, sitotoksik T hücrelerinden Garanzim B salg›lanmas›; mitokondrial enzimlerin sitokrom C üzerinden indirek uyard›¤› bafllang›ç ve infazc› kazpazlar› direkt olarak uyarmaktad›r. Kaspaz terimi, bu enzim ailesinin ‘‘c’’ (cistein proteaz), ‘‘aspaz’’ (molekülü aspartik asit bölgesinden kesebilme) katalitik özelliklerini gösterir. Kaspaz ailesi hücre ölümündeki yer al›fl s›ras›na göre bafllang›ç (apaf-1 molekülünü ba¤layan kaspaz 9 ile fas-fas ligand iliflkisi ile aktive edilen kaspaz 8) ve infazc› bireylerden oluflmaktad›r. Bafllang›ç kaspaz tetiklenince enzimatik ölüm program› h›zl› ve s›ral› bir flekilde aktive edilir ve infazc› kaspazlar, özellikle kaspaz 3 aktivasyonu ile hücre iskelet proteinleri ile nukleer matriks proteinleri y›k›l›r (1-3,6,23,25,42,43). S›n›rs›z Ço¤alma Potansiyeli: Telomeraz, Telomer ve Kanser Normal flartlar alt›nda hücreler belli bir say›da bölünmeden sonra terminal –art›k bölünmeyen- bir duruma gelir. Bu bir anlamda “gençlik ve bölünebilme – yafllanma ve bölünememe” davran›fl›n›n kromozom uçlar›nda bulunan ‘’telomer’’ isimli, hücrenin bölünmesini ve ömrünü sayan sayaç olarak kabul edilebilecek özel yap›lar sayesinde kontrol edildi¤i düflünülmektedir. Normalde her hücre 60-70 kere bölünebilmekte, daha sonra bölünme kapasitesini yitirerek “yafllanma” faz›na girmektedir; bu olay her bölünmede telomerlerin k›salmas›, belli bir k›salma sonras› ise DNA tamir mekanizmalar› taraf›ndan çift sarmal DNA hasar› olarak alg›lan›p, p53 ve Rb ürünleri arac›l›¤› ile hücre döngüsünün durdurulmas›yla sa¤lan›r. Buna karfl›l›k, germ hücreleri gibi bölünmesi sürekli devam etmesi gereken hücrelerde “telomeraz” enzimi bulundu¤undan telomer k›salmas› ile bafl edilebilmekte, hücrelerin ard›fl›k flekilde ço¤almas›na imkan sa¤lanmaktad›r. Telomerlerin k›salmas›na ra¤men hücrenin ço¤almaya devam etmesi, kromozom insta- BÖLÜM 19 ❏ Kanserin Moleküler Temeli ve Jinekolojik Kanserlerde Önemi bilitesini art›rarak tümor progresyonuna katk›da bulunur. p53 ya da Rb mutasyonlar› ile hücre döngüsü kontrol noktalar›n›n inaktif hale getirildi¤i durumlarda hücreyi korumak için kromozomlar›n k›sa uçlar› ucuca eklenerek hücre kurtar›lmaya cal›fl›l›r, ancak bu hatal› tamir sistemi daha da yeni DNA hasarlar›na yol açar ve “kromozom instabilitesi” ortaya ç›kar. Di¤er bir tabirle, tümör hücresinin sonsuz ço¤almas› için büyüme kontrolunun kayb› yeterli de¤ildir, ayn› zamanda hücre yafllanmas› ile bafl edilmesi ve kromozom instabilitesi de gereklidir. E¤er kriz an›nda hücre telomeraz aktivitesini sa¤layabilirse ölümden kaçabilir, ancak telomeraz aktivasyonu öncesi genomik instabilite s›ras›nda çok say›da mutasyonun birikmesi nedeni ile hücre maligniteye ilerlemekten kaçamaz. Kanserlerin %80’inden ço¤unda anormal telomeraz aktivitesi sonucu oluflan bir telomer korunmas› vard›r, baz› kanserlerde ise DNA rekombinasyonu arac›l›¤› ile oldu¤u düflünülen alternatif telomer uzama mekanizmalar› bildirilmektedir, böylece tümör hücreleri uzun yaflar ve daha çok bölünebilir (1-3,6,20,21,23, 44). Tümör Damarlanmas› : Anjiogenezis Yukarda sayd›¤›m›z tüm anormallikler mevcut olsa da, e¤er solid bir tümörün damarlanmas› yoksa 1-2mm den daha fazla büyümesine imkan yoktur. Gerek oksijen ve besin, gerekse at›klar›n diffüzyon yoluyla geçmesi maksimum 1-2mm mesafeye hitabetti¤inden normal dokularda oldu¤u gibi, tümörler de oksijen ve besin sa¤lamak ile at›klar›n at›l›m› için damarlanmaya ihtiyaç gösterir. Damarlanma hem tümör büyümesi hem de metastaz oluflumunda çok önemlidir, damarlanmas› iyi olmayan tümör nekroza gider. Bununla birlikte bu damarlanma, normalde gördü¤ümüz damarlanmadan farkl›l›klar gösterir: damar duvarlar› kuvvetli de¤ildir, afl›r› geçirgendir, dilatedir, iliflkileri rastgeledir (2,6,45). Damarlanma sadece hücre beslenmesini sa¤lamakla kalmaz, endotelden sal›nan çeflitli büyüme faktörleri sayesinde de tümör büyümesi indüklenir. Damarlanma için gerekli faktörler hem bizzat tümör hücrelerinden hem de makrofajlar ve di¤er iltihap hücrelerinden salg›lanmaktad›r. Bu faktörler aras›nda özetle insulin benzeri büyüme faktörü (IGF), PDGF say›labilir. Tümör büyümesinin erken dönemlerinde damarlanma yoktur ve tümör in situ olarak y›llarca kalabilir. Anjiogenik faktörlerin sal›n›m› ya da inhibitörlerin ortadan kalkmas› ile damarlanma bafllar, bu faktörler tümör hücrelerinin kendisi yan›s›ra stromal hücreler ve makrofajlardan da salg›lanabilir. Pek çok proteaz ekstrasellüler matrikste depolanan bFGF salg›layabilirken, potent angiogenez inhibitörleri olan angiostatin, vaskülostatin ve endostatin s›ras› ile plazminojen, kollagen ve transthyretin parçalanmas› ile ortaya ç›kmaktad›r. Oksijenin nisbi olarak biraz da olsa azalmas› oksijene hassas bir transkripsiyon faktörü olan HIF-α (hipoksinin indükledigi gen) ortaya ç›k›p VEGF ve bFGF gibi çesitli angiogenik sitokinlerin salg›lanmas›n› aktive eder. Bu faktörler tümöre do¤ru endotel büyümesini uyar›r. VEGF ayn› zamanda dolayl› olarak Notch sinyal yolunu da uyararak yeni damarlar›n dansitesini ve dallanmas›n› da regüle eder. 11 Gerek angiogenik gerekse antianjiogenik faktörler, kanserde s›kl›kla mutasyon gösteren genler tarafindan regüle edilmektedir: Mesela normal hücrelerde mutant olmayan p53, antianjiogenik bir molekül olan trombospondin 1’i indükleyerek ya da VEGF gibi anjiogenik molekülleri bask›layarak angiogenezi önler. Bu nedenlerle yukar›da bahsedilegelinen p53 özelliklerine ilaveten, burada da p53 kayb›n›n angiogenezis için uygun bir ortam oluflturdu¤u görülmektedir. Ayr›ca VEGFin mutant RAS-MAPkinaz yolundan da indüklenebildi¤i anlafl›lm›flt›r. bFGF ve VEGF tümörlerde s›kl›kla eksprese olduklar›ndan BEVACIZUMAB isimli monoklonal anti-VEGF antikoru son zamanlarda kullan›ma girmifl ve çeflitli kanserlerde kullan›lmaya bafllam›flt›r. Ayr›ca NOTCH aktivasyonunu inhibe eden bir baflka antikor stratejisi daha kabullenilmifltir (1-3,21,23,31,39). Tümör ‹nvazyon ve Metastaz› Metastaz, tümör hücrelerinin primer tümörden farkl› bir bölgede yerleflip ikincil tümör oluflturmas›d›r. Bu özellik benign tümörleri malign tümörlerden ay›ran en önemli fark olup çok basamakl› tümör progresyonunun sonra gelen ama önemli basamaklar›ndand›r. ‹nsan tümörlerinin ço¤u primer neoplazm›n etkilerinden dolay› de¤il, yayg›n metastazlar nedeni ile ölüme neden olmakta ve metastaz, tedavi baflar›s›zl›lar›n›n baflta gelen nedenini oluflturmaktad›r. Asl›nda metastaz primer tümörden hücrelerin vücut boflluklar›na dökülmesi yoluyla da olabilmektedir ama metastaz denince ço¤unlukla akla lenfatik yolu ile lenf nodlar›na ya da kan yolu ile vücut organlar›na olan yay›lma gelir. Primer tümörden milyonlarca hücre dolafl›ma geçse bile gerçekte bu hücrelerin sadece çok az› metastaz yapabilme kapasitesine sahiptir. Metastaz, meme kanserlerinde s›kça görüldü¤ü gibi primer tümörün cerrahi rezeksiyonundan y›llarca sonra dahi ortaya ç›kabilir, ama kanser hastalar›n›n pek ço¤unda tan› s›ras›nda metastaz mevcuttur; bu da ‘kötü prognoz’ ile efl anlaml›d›r. Bu nedenle metastatik potansiyeli belirleyen parametreler klinik olarak son derece önemlidir (2,20,21,46-50). Metastaz Geneti¤i Tümör hücresinin metastaz yapabilmek için di¤er hücrelerden ayr›lmas›, ekstrasellüler matrikse tutunmas› ve onu parçalayarak ilerlemesi, bunun için yeterli hareket (migrasyon) kapasitesine sahip olmas›, damar veya lenfatik içerisine girdi¤inde veya dokuda konakç› immün sisteminden kaçmak ya da korunmak için yeterli korunmaya sahip olmas› gerekir. Kanser metastaz›na yol açan olaylarla ilgili yukar›daki özetten de anlafl›labilece¤i gibi hücreleraras› ve hücre-matriks adhezyonunda, motilitede de¤ifliklikler oluflturabilen, çevre dokuya invazyon, lenfatik ve vasküler yap›lara ulafl›m sa¤layan, bu bölgelerde yaflam› sürdürme ve konakç› defans mekanizmalar›na karfl› koyma ya da sekonder bölgelerde konaklama ve yeni kitleler oluflturma için gerekli genlerin aktivasyonu ile oluflur (6,46-49). ‹lgili olaylar›n detay› ve iliflkili genlerin pek ço¤u afla¤›da özetlenece¤i üzere bugün için anlafl›lmakla birlikte mikrometastazlar›n önceden tayini ve metastatik hastal›k için seçici tedavi uygulan›m› hala onkolojinin önemli problemleri aras›nda yer almaktad›r (20). 12 J‹NEKOLOJ‹K ONKOLOJ‹ Metastazla ilgili genler, metastaz evreleri göz önüne al›narak anlat›lmaya çal›fl›lacakt›r. Tümör henüz lokalize iken, yukarda detayl› olarak anlat›ld›¤› üzere, önce hücre transformasyonu ve sonra transforme hücrenin ço¤alarak büyümesi aflamalar›ndan geçer. ‹nvazyon ve metastaz için neoplastik hücrelerin di¤erlerinden farkl› özellikler tafl›mas› flartt›r. Primer tümördeki hücrelerin sadece bir k›sm›n›n bu ilave mutasyonlar› tafl›yabilece¤i aç›kt›r. Hücreleraras› ‹liflkiler Öncelikle metastaz yapacak bu hücrelerin di¤erlerinden kopabilmesi gerekir –metastatik hücre hücreleraras› adhezyonun azalmas› ile di¤erlerinden ayr›labilmeli, daha sonra hücreleraras› matriks içerisinde ilerleyebilmek için matrikse ba¤lanma ve ve onu parçalayabilme özelliklerine sahip olmal›d›r. Hücreleraras› iliflkiler kadherin ailesi taraf›ndan sa¤lan›r, epitel hücreleri aras›nda yerleflen E-kadherinler hücre içinde β-katenin ve aktin iskeletine ba¤lan›r. Endometrioid endometrium ve over kanserlerinde, baz› kolon ve meme adenokanserlerinde oldu¤u gibi Ekadherin ekspresyonu azal›r ve hücreleraras› ba¤lant› gevfler. Ayr›ca E-kadherin hücre iskeletine kateninler arac›l›¤› ile ba¤lanarak fonksiyon gördü¤ü için E-kadherin bozulmasa da katenin gen mutasyonlar› varl›¤› ayn› sonuca götürmektedir (2). Ekstraselluler Matrikse ‹nvazyon Tümör hücrelerinin hücreleraras› matriks proteinlerine ba¤lanmas› ve polarizasyonu da bozulmufltur: bu ifllev, bazal membranda bulunan laminin ve fibronektine ba¤lanan integrinler sayesinde baflar›l›r. Normal flartlar alt›nda hücreleraras› adhezyon kayb› hücreyi apoptoz ile ölüme götürmekte ise de tümör hücreleri bu tip ölüme rezistand›r. ‹nvazyon için bazal membran ve hücreleraras› ba¤ dokusunun parçalanmas›, ya tümör hücreleri taraf›ndan direk olarak salg›lanan, ya da tümör hücrelerinin uyarmas› ile stromal/iltihabi hücrelerden salg›lanan proteolitik enzimler sayesinde olmaktad›r. Ekstrasellüler matriks, bazal membrandaki kollajen tip IV’ü parçalayan matriks metaloproteinazlar (MMP), katepsin D, ürokinaz plasminogen aktivatorü gibi moleküller arac›l›¤› ile olur. Bahsedilen basamakta sadece interstitsyel matriksin y›k›lmas› de¤il, ayn› zamanda ekstrasellüler matriksin parçalanmas› ile kemotaktik – angiogenik ve büyüme üzerine pozitif etkileri olan faktörlerin de a盤a ç›kmas› söz konusudur (6,51). Metastatik hücre böylece bazal membran› parçalar, “migrasyon” ad› verilen yine kompleks ve birçok sinyal molekülü / reseptörünü içeren çok basamakl› bir süreçle, parçalanan bazal membran ve proteolize u¤rayan hücreleraras› matriksten lenfatik ve vasküler yap›lara gelip, duvarlar›n› yukar›daki ekstrasellüler matriks parçalamas›na benzer bir biçimde parçalayarak dolafl›ma ulafl›r. land›r›lan apoptoz yan›s›ra immün mekanizmalar gibi çeflitli yollarla ortadan kald›r›lmaya çal›fl›l›r. Bununla birlikte tümör hücreleri kendi aralar›nda (homotipik) ve kan hücreleri-trombositler ile (heterotipik) agregatlar oluflturarak yaflamlar›n› uzatmaya çabalar, koagülasyon faktörlerine ba¤lan›p aktive ederek emboli oluflturabilir. Varaca¤› hedefte metastaz›n ilk basamaklar›ndakine benzer flekilde integrin, laminin reseptörleri gibi adhezyon molekülleri ve proteolitik enzimlerin yard›m›yla endotele tutunarak bazal membran› aflar (1-3). Metastaz için seçilen organ genellikle primer tümörün tipi ve yerleflimine ba¤l›d›r ve ço¤unlukla dissemine olan hücreler anatomik yol izler, di¤er bir deyimle, karfl›lafl›lan ilk kapiller yatak veya lenfatik yolu ile olur. Lenf nodlar›na ulaflt›¤›nda tümör hücreleri subkapsüler sinüslerde retiküler fibrillere laminin, fibronektin, kollagen IV gibi yukar›da bahsedilen invazyon s›ras›nda ifllevi olan ekstrasellüler matriks molekülleri ile tutunur. Bununla birlikte metastazda bronflial kanserin adrenallere, beyine; meme kanserinin karaci¤ere, kemik, overlere, prostat kanserinin kemi¤e ya da melanomun beyine olan metastazlar› gibi -yukar›da bahsedilenden farkl› metastaz yollar› için- Stephan Paget 1889’da ‘seed and soil’ (‘tohum ve toprak’) hipotezini ortaya atm›flt›r: özel kanser hücreleri (tohum) baz› özel organlarda yerleflmeye e¤ilimlidir (toprak). Metastaz olacak organ uygun olmal›, yeterli miktar ve uygun tipte büyüme faktörleri içermeli, tümör hücrelerini ‘y›k›c›’ proteaz inhibitörleri bak›m›ndan fakir olmal›d›r. Organ tropizmi için tümör hücreleri hedef organ endotellerine has adhezyon molekülleri eksprese edebilir, hedef organ taraf›ndan baz› kemoatraktif maddeler sal›nabilir ya da tam tersine, baz› organlar metastaz için uygun olmayan özellikte maddeler içeriyor olabilir (46). Endotele tutunmada üzerinde en çok çal›fl›lan molekül, normalde T lenfositlerin görev yapaca¤› bölgeye gitmesinde rolü olan CD44 molekülüdür ve prostat, larinks, serviks ve endometrial kanserlerinde önemli prognostik rolü mevcuttur (13,52-54). Neden her tümör metastaz yapmaz? Metastaza neden olan gen de¤iflikleri nelerdir? sorular› henüz aç›klanabilmifl de¤ildir, “klonal evolüsyon”, “metastatik signatür” modelleri, konakç›n›n genetik zemini veya “kök hücre” hipotezleri ile aç›klanmaya çal›fl›lmaktad›r (46-48,50,55). Bugün için tek bir ‘metastatik’ ya da ‘metastaz bask›lay›c›’ gen tariflemek mümkün de¤ildir, yukar›da anlat›lmaya çal›fl›lan metastaz olay›n›n çeflitli basamaklar›nda yer alan moleküller (adhezyon molekülleri, reseptörleri, kollagenazlar, antikollagenazlar, motilite faktörleri, tümör antijenleri) metastaz ile ilgili genler olarak düflünülmelidir ancak bu genlerin ekspresyonunun zamanlanmas› tümör hücresinin yaflam› için son derece önemlidir. Metastaz bask›lay›c› oldu¤u iddia edilen genler Tablo 19-5’de özetlenmifltir (3,6,20,39,46-48,55-59). Vasküler Da¤›l›m, Yerleflme (homing) ve Kolonizasyon Kanserde Gen Regülasyonu Bozukluklar›, Genomik ‹nstabilite, DNA Tamir Bozukluklar› Dolafl›ma giren tümör hücresi; mekanik bas›nç, hücreleraras› adhezyon kayb› nedeni ile indüklenen ve ANOIKIS olarak ad- Gerçekte çevremizde çok yo¤un bir DNA zedeleyici etken okyanusu bulunmas›na ra¤men kanserin her bireyde görülüyor BÖLÜM 19 ❏ Kanserin Moleküler Temeli ve Jinekolojik Kanserlerde Önemi TABLO 19-5 13 Muhtemel Metastaz Bask›lay›c› ve ‹ndükleyici Genler Bask›lay›c› Gen ‹lgili Kanser Mekanizma nm23 (NDPK) ailesi PTEN E-Kadherin MK4 KiSS-1 BRMS 1 DPC4 Mikro RNA 335 ve 126 Meme, kolon,larinks, serviks, melanom Prostat, glioma, meme Pek çok adenokarsinom Prostat Melanom, meme Meme Kolorektal, pankreas Meme G protein sinyal, mikrotübül birleflmesi Migrasyon, fokal adhezyon Hücreleraras› adhezyon Strese cevap Sinyal iletimi Hücreleraras› iletiflim ? Gen ekspresyonu de¤iflikli¤i ‹ndukleyici Gen ‹lgili Kanser Mekanizma SNAIL, TWIST Meme Epitelyal-mezenkimal geçifl olmamas›n›n nedeni, organizmada DNA zedelenmesini tan›ma ve e¤er normale dönebilecek bir zedelenme ise de tamir edebilme yetene¤inin var olmas›ndand›r. DNA tamir mekanizmalar›nda kal›tsal defekt bulunan kifliler ya da oluflan sporadik defektler, bu genlerin onkogen niteli¤inde olmamas›na ra¤men hücre bölünmesi ile ilgili genlerde oluflabilecek mutasyonlara öncelik ederler. Asl›nda “genom instabilitesi” için DNA tamir geninin her iki kopyesi de zedelenmifl olmal›d›r, bununla birlikte gen dozaj› ile (di¤er bir deyimle haploinsufficiency) ile kansere neden olan DNA tamir genleri de vard›r. Genelde 3 tip DNA tamir mekanizmas›; “mismatch” repair (yanl›fl eflleflme tamiri), nükleotid eksizon tamiri ve rekombinasyon tamiri fleklinde özetlenebilir. Mismatch tamir geninin ailevi bozuklu¤u durumu Herediter Nonpolipozis Kolon kanseri sendromu (HNPCC); nükleotid eksizon tamiri genlerinde kal›tsal bozukluklar XerodermaPigmentozum (XP), homolog rekombinasyon tamiri bozukluklar› ise Bloom sendromu, Ataksi-Telenjiektazi-Fankoni anemisi örnekleriyle k›saca özetlenecektir. Mismatch Tamir hatalar›: Herediter Non-Poliposis Kolorektal Kanser Ailevi non-polipozis kolorektal kanser sendromu (HNPCC) öncelikle çekum ve ç›kan kolonu ilgilendiren kanserlerle belirir ve DNA yanl›fl eflleflmesini tamir eden genlerin bozukluklar› ile oluflur. Herhangibir DNA dizisinin ço¤almas› gerekli oldu¤u durumlarda, DNA bazlar›n›n do¤rulu¤unu kontrol eder, varsa yanl›fllar› düzeltir. Aksi taktirde genomdaki hatal› ço¤alma, zaman içerisinde birikerek yukar›da bahsedilegelen “kanser oluflumu” genlerini de ilgilendirecektir. Mismatch tamir genleri “mikrosatellit instabilitesi” (2,5) ile karakterlidir. Mikrosatellitler, bir ila alt› nükleotidden oluflan ve k›saca, genom boyunca rastgele tekrarlayan birimlerdir ve normal bir kiflide uzunluklar› sabit kalmakta olmas›na ra¤men HNPCC ailelerinde genlerin stabil olmamalar› nedeni ile tümör hücrelerinde uzunluklar› artar ya da k›sal›r ve ayn› hastan›n normal hücrelerinde bulunmayan alleller ortaya ç›kar›r. Birçok DNA mismatch tamir geni olmakla birlikte 2p16 yerleflimli MSH2 ile 3p21 yerleflimli MLH1 genlerindeki germline mutasyonlar bu hastalar›n en az›ndan üçte birinde mutlaka mevcuttur. HNPCC sendromu tüm kolon kanserlerinin %5inden az›n› oluflturmakla birlikte, bu genlerdeki de¤ifliklik sporadik kolon kanserlerinin %15-20’sinde görülmektedir. Afla¤›da detayl› anlat›laca¤› üzere sporadik endometrioid tip endometrial kanserler ile endometrioid over kanserlerinde de mutasyon gösterirler. Genetik iflleyifl ve kal›t›m bak›m›ndan DNA tamir genleri tümör bask›lay›c› genlere benzemekle birlikte onlardan farkl› olarak hücre büyümesiyle direkt iliflki göstermezler. Do¤rusunu söylemek gerekirse, HNPCC ailelerindeki defektler, TGF-βRII, β-katenin yolundaki baz› genler, BAX gibi büyümeyi regüle eden di¤er onkogen ve tümör bask›lay›c› genlerde de hatalara neden olur (2,3,5,20,42,60,61). Nükleotid eksizyon tamiri hatalar›: Xeroderma Pigmentozum Günefl ›fl›n›ndaki ultraviyole radyasyonun neden oldu¤u pirimidin çiftlerindeki hatal› ba¤lanma, “nükleotid eksiyon tamiri” ile düzeltilir, ve bu yolakta bulunan proteinlerden herhangibirinin kal›tsal yoklu¤u Xeroderma Pigmentozum hastal›¤›na yol açar (62). Homolog rekombinasyon hatalar›: Bloom sendromu (BS), Ataksi-Telenjektazi (A-T) ve Fankoni anemisi (FA) Bu grup hastal›klar otozomal resesif geçifllidir ve defektli hastalar BS ve A-T’de oldu¤u gibi ya iyonize radyasyon, ya da FA’deki çeflitli kemoterapötikler gibi DNA ba¤lay›p zedeleyici ajanlara afl›r› sensitivite gösterirler. Bu gruptaki hastal›klar›n fenotipleri de oldukça kar›fl›kt›r, kansere predispozisyona ilaveten mesela A-T nöral semptomlar, FA kemik ili¤i aplazisi, BS ise geliflim defektleri ile gider. Daha önce de belirtildi¤i gibi, A-T’de mutant olan ATM geni iyonize radyasyonun oluflturdu¤u DNA hatas›n› tan›y›p tamir eden bir gendir. BS ilgili gen 15.ci kromozomda yer al›r ve homolog rekombinasyon ile oluflan DNA tamirinde görev yapar. Fankoni anemisi gen kompleksinde 13 ayr› gen vard›r ve herhangi birindeki defekt FA hastal›¤›na neden olur. Konumuzla çok ilgili olarak BRCA2 geni de bu hastalar›n bir k›sm›nda defektlidir (63). 14 J‹NEKOLOJ‹K ONKOLOJ‹ Epigenetik De¤ifliklikler ve Metilasyon Genetik de¤iflikliklerin ço¤u tümör gelifliminin belirli evrelerine has iken; epigenetik de¤ifliklikler, mutasyon olmadan gen ekspresyonunda oluflan kal›tsal ve geri dönüflümlü de¤ifliklikleri simgelemektedir. DNA metilasyonu ve histon genlerinde translasyon sonras› olan modifikasyon, gen ekspresyonunu etkileyen en önemli epigenetik de¤iflikliklerdir. Normalde memeli genomundaki farkl›laflm›fl somatik hücrelerde genomun büyük bir k›sm› eksprese olmaz, 5’-CpG-3’ dinükleotidlerinin bir k›sm›nda oluflan DNA metilasyonu ile genomun %80’i metillenmifl, modifiye edilmifl ve sessiz hale getirilmifl olup DNA heterokromatin fleklinde kompaktlaflt›r›lm›fl flekildedir. Ancak kanser hücreleri global bir hipometilasyon ve baz› promotorlarda seçici olarak hipermetilasyon ile karakterlidir. Kolorektal kanser hücrelerinde ve adenomatöz poliplerde DNA metilasyonunda azalma bulunmufltur (3,22,64). Son y›llarda baz› tümör bask›lay›c› genlerde mutasyon olmadan, promotorlar›nda oluflan hipermetilasyonla inaktif hale getirildikleri anlafl›lm›flt›r. Buna en güzel örnek, iki tane tümör bask›lay›c›y› p14/ARF ve p16/INK4A’y› kodlayan CDKN2A’da oland›r: p14 kolon ve mide kanserlerinde, p16/INK4A çeflitli kanserlerde epigenetik olarak susturulmufltur. CDKN2A loküsünün kodlad›¤› genler p53 ve RB ile iliflkili olduklar›ndan, tek bir epigenetik de¤ifliklik, iki ayr› tümör bask›lay›c› geni birden inaktive etmeye neden olur. Metilasyonla susturulan di¤er tümör bask›lay›c› genler aras›nda meme kanserinde BRCA1, renal hücreli karsinomda VHL, kolorektal kanserde MLH1 DNA tamir geni say›labilir. Metilasyon ayn› zamanda gen veya kromozomun maternal ya da paternal allelinin metilasyon ile inaktive edildi¤i “genomik imprinting” olay›nda da yer ald›¤›ndan, baz› tümör hücrelerinde tersi bir olay yani demetilasyon ile oluflacak imprinting kayb› sonucu - iki allelin birden ekspresyonu- da söz konusu olabilmektedir. Son y›llarda deney hayvanlar›nda oluflturulan genom hipometilasyonunun, kromozom instabilitesi ve artan tümör insidans›na neden olmas› epigenetik de¤iflikliklerin direk olarak tümör geliflimine katk›s›n› vurgulam›flt›r; bu nedenle, tümör bask›lay›c› genlerde DNA bölgelerini demetile eden potansiyel tedavi ajanlar›n›n gelifltirilmesine yönelik çal›flmalar artm›flt›r (2,3,5,23). Kromatin de¤iflikliklerinin karsinogeneze katk›s› daha az anlafl›labilmifltir, bugünkü bilgilere göre asetilasyon ve metilasyon gibi çeflitli histon modifikasyonlar› transkripsiyonun aktivasyon veya represyonuna neden olur, EZH2 gibi çeflitli kromatin modifikatör enzimleri meme ve prostat kanserleri gibi baz› tümörlerde afl›r› eksprese olmaktad›r. EZH2, hücre kültürlerinde p21 gibi tümör bask›lay›c›lar› represe etmekle birlikte in vivo tümörlerdeki hedefleri henüz anlafl›lm›fl de¤ildir. Ayr›ca kromatin biçimleyici enzimler ile DNA metilasyon mekanizmas› aras›nda da henüz tam olarak bilemedi¤imiz çok önemli iliflkilerin oldu¤u düflünülmektedir (64,65). ÇOK BASAMAKLI “MULT‹STEP” KARS‹NOGENEZ, KOLOREKTAL ADENOMA-KARS‹NOMA ‹LERLEY‹fi‹ Kolorektal karsinogenez, daha önce bahsedilen multipl genetik de¤iflikli¤e iyi bir örnek teflkil eder. Kolorektal kanserin adenomdan geliflti¤ini destekleyen pekçok delil mevcuttur. Kolorektal kanser gelimesi için kabul edilen genetik model fiekil 19.3’de sunulmaktad›r (21). ‹zledi¤iniz genetik model flimdiye kadar bildirilen çeflitli çal›flmalar›n biraraya getirilmesi ile oluflmufltur, ancak tam olmad›¤› ve afl›r› basite indirgendi¤ini kabul etmek gerekir. Ayr›ca bu modelde gen ekspresyonu ya da aktivitesi, büyüme faktörüne cevap, angiogenezis, motilite ve kolorektal karsinom hücrelerini normal kolon epitel hücrelerinden ay›ran ve son y›llarda üzerinde çal›fl›lan pekçok özelli¤in mevcut olmad›¤›n› hat›rlatmak gerekir (5,21-23). fiEK‹L 19-3 Kolorektal kanser genetik modeli. Kolorektal kanserlerin büyük bir ço¤unlu¤unun adenomatöz polip zemininde on y›llar süren bir zaman diliminde gelifltiklerini biliyoruz. Kal›tsal ya da somatik genetik de¤ifliklikler tümör inisiasyon ve progresyonunu etkiler. Bu de¤iflikliklerde s›ralanma fark› olabilse de, tümörigenezisin çeflitli evreleri aras›nda s›k› bir iliflki vard›r (Fearon ER ve Vogelstein B. Cell 61:759,1990’dan modifiye edilmifltir). BÖLÜM 19 ❏ Kanserin Moleküler Temeli ve Jinekolojik Kanserlerde Önemi KÖK HÜCRE, EMBR‹YON‹K KÖK HÜCRE, iPS HÜCRE KAVRAMLARI Güncel bir konu olmas› nedeniyle rejeneratif t›pta ufuk açan ve bilgi sahibi olman›n çok yararl› oldu¤unu düflündü¤ümüz kök hücre konusunda k›saca bilgi vermek istedik. Hücre farkl›laflmas› ile ilgili bilgilerin geliflmesi sayesinde, bu bilgiler kullan›larak kök hücrenin farkl›laflmas› ve sonuçta kalp, beyin, karaci¤er ya da kas gibi zedelenmifl insan dokular›n›n yerini alacak flekilde organize edilmesi, bu dokular›n hastal›klar›n›n tedavisinde kullan›lmas› amac›na dayanmaktad›r. Kök hücreler kendi kendini yenileme ve farkl›laflm›fl hücre klonlar› oluflturma potansiyeline sahiptir. Normalde embriyonik geliflmenin erken evrelerinde ilerde organizmay› oluflturacak kök hücreler embriyonik kök hücre (ES) olarak adland›r›l›r, bilindi¤i üzere embriyonun blastosist evresindeki bu hücreler pluripotenttir (yani pankreatik adac›k hücreleri, hepatositler, nöronlar dahil vücudun her türlü dokusuna farkl›laflma ve kendi kendini yenileme potansiyeline sahiptir). Pluripotent kök hücrelerden türeyen bir sonraki jenerasyon multipotent kök hücreler olup, geliflim potansiyelleri daha s›n›rl›dr; multipotent kök hücrelerden de, en sonra endoderm/mezoderm ya da ektoderm embriyonik tabakalar›na ait farkl›laflm›fl hücreler geliflir. Eriflkinde ise kök hücreler genellikle ER‹fiK‹N KÖK HÜCRE ya da SOMAT‹K KÖK HÜCRE olarak adland›r›lan hücreler olup pek çok dokuda farkl› hücre tiplerini oluflturma kapasitesine sahiptir. Deri, intestinal mukoza, kornea ve özellikle kemik ili¤i dokular›nda bu tip hücrelerin mevcut oldu¤u son y›llarda anlafl›lm›flt›r. Eriflkin insan ve hayvanlar›n merkezi sinir sistemi dokular›nda bile kök hücre varl›¤› saptanmas› hayret uyand›rm›flt›r. 2006’da fareden ve 2007’de insan hücrelerinden türetilen “Induced pluripotent kök hücreler” (iPS veya iPSCs) sayesinde araflt›rmac›lar art›k hem embriyo kullanmadan pluripotent hücre yapabilme flans›na sahip olmufl, hem de hastan›n bizzat kendi hücrelerinden geliflirildikleri için tedavi amaçl› olarak hastaya geri verildiklerinde graft vs host ya da immün at›l›m gibi problemlerin de ortadan kalkmas›na neden oldu¤undan 盤›r aç›lm›flt›r. Bu geliflimi k›saca basamak basamak özetleyecek olursak, önce eriflkin dokular›n farkl›laflm›fl hücrelerinden çekirdekleri al›narak nükleusu ç›kar›lm›fl bir oosite transferi ve bu melez oositin daha sonra do¤uracak kiral›k anneye (surragate mother) transfer edilmesi ile “reproduktif klonlama” 1997 y›l›nda koyun Dolly örne¤inde baflar› ile yap›labilmiflti (2). Bu geliflme sonras›nda ayn› teknik kullan›larak “terapötik klonlama” ve insan hastal›klar›n›n tedavisi konusunda umutlar artm›flt›r. Bu teknikte ise k›saca, hastaya ait insan fibroblast çekirdekleri, nükleusu olmayan insan oositine verilerek yaflam kayna¤› baz› genlerin transferi sayesinde pluripotent olmalar› sa¤lanan ES hücreleri oluflturulmakta, kiral›k anne karn›nda de¤il de kültürde ço¤alt›labilen bu hücrelerin de daha sonra kas, nöron, hematopoietik hücre gibi de¤iflik hücrelere farkl›laflmas› sa¤lan›p, prensip olarak sonradan kültür ortam›nda is- 15 tenildi¤i kadar ço¤altilabilen bu dokular›n hastaya geri verilmesi ile zarar görmüfl organlar›n yeniden oluflumu hedeflenmektedir. iPS hücrelere embriyosuz ES hücreler de denmektedir. Bununla birlikte tekni¤in güçlü¤üne ilaveten hem reproduktif hem de terapötik klonlaman›n yeniden programlanan ES hücrelerinde histon metilasyon yetmezli¤i ve bozuk gen ekspresyonu gibi baz› genetik de¤iflikliklere yol açmas› en önemli baflar›s›zl›k nedenlerindendir. Pek çok çal›flma sonras›nda ES hücrelerinin pluripotent olmas›n› sa¤layan genlerin 4 önemli transkripsiyon faktörü (Oct3/4, Sox2, c-myc ve Klf4) oldu¤u ve farkl›laflmay› önleyici genin de Nanog ismi verilen “ebediyet geni” oldu¤u anlafl›lm›fl ve kan›tlanm›flt›r. Eriflkin veya yenido¤an insan matür fibroblastlar› bu genler ile indüklenerek yeniden programlan›p iPS hücreleri oluflturularak, hem endodermal, hem mezodermal hem de ektodermal orijinli hücrelerin elde edilebilece¤i ve orak hücre anemi modeli fareyi tedavi etmekte kullanabilece¤i gerçe¤i, genetik manipulasyon ve sonra da transplantasyona ra¤men hücrelerin fonksiyon görmeye devam ettiklerini kan›tlamaktad›r. K›saca iPS hücreler oositlere nükleer transfer gere¤i kalmadan, hastalara “kendilerine ait, kendilerine has kök hücre tedavisi” ümidi vermektedir. iPS hücrelerinin insan rejeneratif t›bb›nda kullanabilme rüyas›n›n gerçekleflmesi, yeni gen aktarma metodlar›n›n gelifltirilmesi ve transfer edilen indükleyici genler aras›nda asl›nda onkogen olan c-myc ve Kfl4 genlerinin replase edilebilmesini gerektirmektedir (2, 65a). iPS hücreler embriyonik kök (ES) hücreler gibi pluripotent hücrelerle gen ve protein ekspresyonu, kromatin metilasyon paterni ve farkl›laflma kapasitesi dahil pek çok yönden benzerlik göstermekte olsa da henüz gerçek ve do¤al pluripotent embriyonik hücrelerle olan iliflkileri araflt›rma evresinde oldu¤u bildirilmektedir. ‹laveten, daha önceki iPS metodlar›nda genlerin orijinal hücreye virüsler arac›l›¤› ile transfer edilmesi hedef hücrelerde kanser oluflturma tehlikesi yaratmakta iken, daha sonra kendi genlerini hedef hücre genlerine integre etmeyen adenoviruslerin kullan›m› bu tehlikeyi azaltm›fl, hatta etkinli¤i çok daha düflük olsa da virus kullan›lmaks›z›n plazmidler arac›l›¤›yla bu ifllemin yap›labilece¤i öne sürülmüfl; son olarak da 2009’da genetik modifikasyon için virüslere ihtiyaç olmaks›z›n iPS hücrelere direk protein verilerek pluripotent olmalar›n›n sa¤lanabilece¤i bildirilmifltir. Maalesef transfer edilen genlerden özellikle c-myc geni asl›nda onkogenik oldu¤undan, bir sonraki generasyondaki farelerde %20 kanser geliflmesi baz› araflt›r›c›lar tarafindan iPS hücrelerin “insanlar tarafindan oluflturulan kanser hücreleri” olarak isimlendirilmelerine neden olmufl ve bu tekni¤e karfl› gruplar ortaya ç›km›flt›r (65b). En son bir geliflme olarak da yine Japonya Kyoto grubu, matür hücrelerden oluflturulan hibrid hücrelerin önce geri farkl›laflmas› (dediferansiasyon) ile pluripotent hale getirilmesi ve daha sonra da istendi¤i yönde farkl›laflt›r›lmas›na gerek kalmaks›z›n direk olarak arzu edilen hücreye diferansiye edilebilece¤i fleklinde dikkat çekici raporlar da bildirilmektedir. Çok popüler olan bu konuyu k›saca bu kitapta özet olarak da olsa sunmak istememizin nedeni, art›k günümüzde, insan 16 J‹NEKOLOJ‹K ONKOLOJ‹ diferansiye hücrelerinin, yukar›da sözü geçen baz› transkripsiyon faktörleri ile indüklenip yeniden programlanabilece¤inin anlafl›lmas› ve bu ifllemin teknik olarak gerçeklefltirilebilmifl olmas›d›r. ‹ndüklenmifl pluripotent hücreler (ya da iPS hücreleri) olarak adland›r›lan bu hücrelerin keflfinin, kök hücre araflt›rma ve uygulanmas›nda 盤›r açmas› beklenmektedir. Ayr›ca, kanserin ölümsüz/sonsuz ço¤alma kapasitesine sahip hücrelerden oluflmas›, baz› kanser hücrelerinin “kök hücre” özelliklerini tafl›d›¤›n› düflündürdü¤ünden, kanser dokusunun içindeki kök hücrelerin saptanmas› ve o hücrelerin eliminasyonunun kanserde kür sa¤lama umidini de uyand›rmaktad›r. Mevcut kanser tedavilerinin baflar›lar›n›n s›n›rl› oluflu k›smen de olsa, kanseri oluflturan malign kök hücreleri ortadan kald›rmaktaki baflar›s›zl›¤›m›za ba¤l› olabilir. J‹NEKOLOJ‹K ORGAN KANSERLER‹NDE MOLEKÜLER ÖZELL‹KLER Bu k›s›mdan itibaren, spesifik olarak jinekolojik organ kanserleri tek tek incelenecek; yukar›da anlat›lan mekanizma ve moleküllerdeki de¤iflilikler organ baz›nda özetlenecektir. Amac›m›z sadece prognostik parametrelerle iliflkiyi incelemek de¤ildir, bu moleküllerin karsinogenezde oynad›klar› roller de, prognostik anlamlar› olmasa bile son derece önemlidir. Moleküler belirteçlerin jinekolojik kanserlerdeki durumunu inceleyen çal›flmalar›n ço¤u k›s›tl› hasta üzerinde yap›lan, ve multivaryant analizden yoksun olan, ya da multivaryant analize tüm prognostik de¤iflkenleri dahil etmeyen de¤erlendirmelerdir. Bu nedenle afla¤›da bildirilen iliflkilerin ço¤unun, sadece bizlere önbilgiler veren sonuçlar olarak de¤erlendirilmesi uygun olur. Bu bölümde, elinizdeki kitab›n onceki bask›s›nda anlatt›¤›m›z bilgilerin ço¤unu tekrarlamay›p öncelikle en yeni bilgileri özetlemeyi ye¤ledik. Serviks Kanseri: Karsinogenez ve Moleküler Faktörler Epidemiyolojik çal›flmalar serviks tümörigenezisinde seks yoluyla geçen bir ajan› etyolojik faktör olarak göstermifl, 1960 ve 70’li y›llarda özellikle HSV-2 sorumlu tutularak incelemeler yap›lm›fl, moleküler çal›flmalar HSV-2 ve serviks kanseri aras›ndaki iliflkiyi gösteremeyince HSV’ye olan ilgi kaybolmufltur. 1976’da Zur Hausen baz› human papilloma virus (HPV) tiplerinin serviks tümör oluflumu ile ilgili olabilece¤ini ileri sürdükten sonra epidemiyolojik çal›flmalarla desteklenen bu bulgu, son zamanlarda moleküler biyolojik çal›flmalarda kan›tlanm›fl, Zur Hausen’e 2008’de Nobel ödülü getirmifltir. Sonuç olarak bugün servikal kanserde etyolojik faktör olarak HPV enfeksiyonunu gösteren yeterli dellillere sahibiz. Çift sarmal bir DNA virüsü olan HPV enfeksiyonlar›n›n ço¤u geçicidir ve muhtemelen konakç›n›n hücresel ba¤›fl›kl›¤› ile ilgili olarak, tesbit edildikten 1-2 y›l sonra latent hale gelir ya da kendili¤inden ortadan kaybolur. Papilloma virusleri genelde benign lezyonlarla birlikte olanlar (düflük/intermediate riskli grup: tip 6, 11, 30, 34, 40, 42, 43, 44, 57) ve invaziv kanserlerle birlikte olanlar (yüksek riskli=kanserle iliflkili grup: tip 16, 18, 33, 35, 39, 45, 51, 52, 56, 58) olarak ikiye ayr›labilir. Yüksek riskli HPV enfeksiyonlar›n›n kendili¤inden ortadan kalkma ihtimali, düflük risklilere göre az ve süresi de yavaflt›r; 36 aydan daha uzun sürerse de ihtimal hemen tamamen ortadan kalkar; üstelik enfeksiyonun uzamas› oran›nda lezyonun yüksek dereceli olma ihtimali artar. Benign lezyonlarda virus genomu serbest ve sirküler nitelikte iken invaziv kanserlerde sirküler formdan epizomal hale geçer ve konakç›n›n serviks hücresi DNA’s›na, ço¤unlukla myc onkogeni bölgesine integre olur. Viral genomda ba¤lanma bölgesi ise E6 ve E7 genlerini regüle eden E2 k›sm›ndand›r. Onkogenik HPV’ler taraf›ndan kodlanan E6 ve E7 proteinleri normal keratinositleri immortalize etme yetene¤ine sahiptir. Her ne kadar HPV ile anogenital neoplazi iliflkisi flüphe götürmeyecek kadar belirgin ise de, tam bir transformasyon için bu proteinlerin yeterli olmayabilece¤i, serviks kanseri geliflmesinde rol oynayan di¤er moleküler olaylar›n varl›¤› ve birikimi gerekti¤i unutulmamal›d›r; telomeraz aktivasyonu ve diger kromozom anormallikleri gibi ilave hasarlar da genellikle HPV integrasyonu ile iliflkilidir. Afla¤›da bahsedilecek ilave moleküler mekanizmalar sadece serviksin epidermoid karsinomalar› için de¤il, aksi belirtilmedikçe yüksek riskli HPV ile enfekte tüm hücre tiplerinden oluflan kanserler için geçerlidir. HPV partiküllerinin hem in situ hem invaziv serviks adenokarsinomalar›nda genoma entegre olarak bulundu¤u gösterilmifltir (68). HPV alt tipinin prognoz üzerindeki etkisi tart›flmal› bir konudur. Baz› çal›flmalara göre HPV 18 kötü prognoza neden olur: 5 y›ll›k hastal›ks›z yaflam›n intermediate tipler ile enfekte olan kanserlerde %100 iken HPV 16 enfeksiyonu ile oluflan serviks kanserlerinde %58, HPV 18 enfeksiyonu ile oluflan serviks kanserlerinde ise %38 oldu¤u bildirilmifl olmakla birlikte konu hala tart›flmal›d›r. C-erbB2 geni ampfilikasyonu ve afl›r› ekspresyonu serviks kanserinde %76’ya varan s›kl›kta olmakla birlikte meme kanserinde kullan›lan standart Hercep Test skorlama kriterlerine göre pozitifli¤i nadirdir. C-erbB2 için immünohitokimya pozitifli¤i di¤er kanserlerde oldu¤u gibi serviks kanserinde de kötü sa¤kal›m ile iliflkili bulunmufl ve yüksek riskli hastal›k ölçeri bir de¤iflken oldu¤u özellikle adenokanser için bildirilmifl, radyoterapi ile tedavi edilen hastalarda da rekürrens ile iliflkili oldu¤u gösterilmifltir. HGF ve reseptörü c-met, hücre proliferasyonunu sa¤layan ve tümör gelifliminde rol alan etkenlerdir. Skuamoz Hücreli Karsinom Antijeni (SCCAg)’nin serum düzeylerinin kötü sonuçlar› öngördü¤ü bulunmuflsa da multivaryant analizde ba¤›ms›z de¤iflken de¤ildir. EGFR ailesinden bir büyüme faktörü olan cripto (CR-1) serviks kanserinde PI3K ba¤›ml› bir sinyal yola¤› üzerinden etkiyerek sa¤kal›ma etkili bir ajan olarak bildirilmifltir. Kendi çal›flmam›zda her ne kadar ‹HK ile saptanan cripto afl›r› ekspresyonu birçok kötü prognostik de¤iflkenle iliflkili bulunmufl ise de multivaryant analizde önemini kaybetmifltir. HGF/c-met kompleksinin afl›r› eksprese olmas› kondiloma akuminatada hücre proliferasyonuna yol açmakta, onkogenik HPV varl›¤›nda ise serviks kanserinde tümör geliflimine neden olmakta, ayr›ca onkojenik HPV’ler ve HIV enfeksiyonu ile güçlü ba¤›nt› göstermektedir. Afl›r› c-met ekspresyo- BÖLÜM 19 ❏ Kanserin Moleküler Temeli ve Jinekolojik Kanserlerde Önemi nunu immünhistokimyasal olarak de¤erlendirdi¤imiz cerrahi evre I ve II olan 94 serviks kanserinde, bu özelli¤in primer tümörün çap›, derin servikal stroma invazyonu, metastatik lenf nodu varl›¤› ve metastatik lenf nodu say›s› ile iliflkili olup multivaryant analizde hastal›ks›z ve 5 y›ll›k sa¤kal›m için ba¤›ms›z prognostikatör oldu¤u bulunmufltur. Bu prognostik önemin HGF’nin VEGF ve tümör anjiogenezis faktörü ile olan iliflkisinden de kaynaklanmas› olas›d›r (10-15, 69-70). EGFR inhibitörlerinin tedavide kullan›m›na gelince, Gefitinib kullan›lan bir faz 2 cal›flmada ‹HK ile saptanan EGFR ekspresyon seviyeleri ile tümör cevab› ve hastal›k kontrolu aras›nda iliflki olmad›¤›n› görmekteyiz (71). EGFR sinyalleri arac›l›¤›yla HPV16’n›n E6 ve E7 proteinleri taraf›ndan regüle edilen, arakidonik asidin prostoglandinlere dönüflmesinde anahtar enzim olan COX-2 ekspresyonu; nonsteroid antiinflamatuvar bir ilaç olan celecocib ile azalmakta, dolayl› olarak da tümörde proliferasyon ve angiogenez azalmakla birlikte; klinik cal›flmalarda bu ilaç radiosensitizer olarak umutsuz sonuçlar vermifltir (72). CIN3’de ve invaziv serviks kanserinde amplifikasyon ve/veya rearranjman ile baz› çal›flmalarda kötü prognozla korele oldu¤u bildirilen c-myc afl›r› ekspresyonu %20 oranda görülür ve genelde HPV16 ile birliktedir (66,73). Di¤er kanserlerde oldu¤u gibi serviks karsinogenezinde de tümör bask›lay›c› genlerin rolü vard›r, daha önce bahsedildi¤i gibi tümör bask›lay›c› genlerin inaktivasyonu (genellikle rearranjman, delesyon veya mutasyon gibi gen seviyesinde de¤iflikliklerle olmakla birlikte), tümör bask›lay›c› gen ürünleri olan proteinlerin inaktivasyonu da söz konusu olmaktad›r: Yüksek riskli HPV tipleri olan 16 ve 18’in E6 proteini spesifik olarak p53 tümör bask›lay›c› gen ürününe ba¤lanmakta ve parçalayarak etkisini yok etmektedir. Benzer flekilde E7 proteini Rb tümör bask›lay›c› gen ürününün fonksiyonunu ortadan kald›r›r, proteini inaktive eder. Yüksek riskli HPV ile enfekte olan hücrelerde, enfekte olmayanlara göre bir büyüme avantaj› olaca¤› aç›kt›r. Gerçekten de serviks kanserleri incelendi¤inde HPV ile enfekte tümörlerde p53 ve Rb genlerinde mutasyon saptanmazken, etyolojide HPV suçlanmayan tümörlerde p53 ve Rb genlerinde mutasyon varl›¤›n›n saptanmas›, bu teorinin geçerlili¤ini ispatlamaktad›r (66,67). p53 geninin 72. pozisyonunda prolin ya da arjinin bulunmas›n› belirleyen s›k bir polimorfizmin, p53 proteininin E6 taraf›ndan y›k›m›n›n arjinin formunda en s›k, heterozigot formda orta düzeyde ve prolin formunda en az oldu¤unun öne sürülmesi ile bu konu dikkatleri çekmifltir. Ancak daha sonraki çal›flmalar bu iliflkiyi gösterememifl ve bu uyumsuzlu¤un 72. kodonun allel tiplendirmesindeki metod farkl›l›klar›ndan kaynakland›¤› yorumu yap›lm›flt›r. p53 bir prognostik faktör olmasa bile, p53’ün inaktivasyonu serviks kanseri gelifliminde anahtar role sahiptir. Bu rol, yukar›da belirtildi¤i gibi onkogenik HPV’ler taraf›ndan üretilen E6 proteininin p53’ü inaktive etmesine ba¤l›d›r. Nadiren görülmekle birlikte p53 genindeki mutasyonlar da CIN’den invaziv karsinoma dönüflümde rol alan faktörlerden olabilir. FHIT geninde heterozigotitenin kayb› (LOH), homozigot delesyonar ve transkript de¤iflimleri CIN ile invaziv karsinom- 17 larda bildirilmifltir, ancak HPV infeksiyonu ya da progresyon ve prognoz ile iliflkisi aç›kl›¤a kavuflmam›flt›r. Segawa E6 ve E7 genlerinin CIN ve invazif karsinom gelifliminde önemli oldu¤unu ancak FHIT ekspresyonu anormal olan hücrelerde kanser geliflimi için E6-E7’ye gerek olmad›¤›n› savunmufltur (41,74). Her ne kadar geçmifl y›llarda mikrodamarlar ile prognoz aras›nda bir iliflki öngörülmemifl olsa da, tümör damarlanmas›n›n en yo¤un oldu¤u alanlarda ortalama damar yo¤unlu¤u (Mikrodamar Dansitesi=MVD) ölçümü sayesinde MVD’nin ba¤›ms›z bir prognostik faktör oldu¤u ve lenf nodu metastaz› ile iliflkili oldu¤u anlafl›lm›flt›r. MVD’nin MMP-9 ekspresyonu dolay›s›yla pelvik ve stromal infiltrayonu, FIGO evresi, histolojik farkl›laflma ve Ki-67 ekspresyonu ile de iliflkili bulundu¤u bildirilmifltir. Ancak bu sonuca ulaflmayan yay›nlar›n da olmas› nedeniyle kullan›lan teknik ve çal›flmay› yapanlar›n tecrübesininin, analiz üzerinde etkili oldu¤u ortaya ç›kmaktad›r. Vasküler Endotelyal Büyüme Faktörü (VEGF), ile prognostik de¤iflkenler aras›ndaki iliflki baz› çal›flmalarda toplam ve hastal›ks›z sa¤kal›m› etkiliyor olarak bulunmuflsa da, bu sonuç tart›flmal›d›r. Yeni çal›flmalar VEGF’ün prognostik önemini anlams›z bulmaktad›r, ancak VEGF-C alt grubu di¤er aile üyelerinin hepsinden farkl› olarak prognoz üzerinde gerçekten etkili olabilir. Bununla birlikte yüksek riskli HPV ile VEGF aras›nda bir iliflki oldu¤u ise aç›k ve tart›flmas›zd›r. HPV 16 E6 onkoproteinin VEGF geninin promoter bölgesini p53’den ba¤›ms›z olarak aktive etmektedir ki bu mutant ras, EGF reseptörü, ErbB2/Her2 ve c-myc’in VEGF ekspresyonunu artt›rmas›na benzerlik gösterir. Rekombinant antiVEGF monoklonal antikoru olan bevacizumab ile yap›lan faz 2 çal›flmalar rekurrent serviks kanserlerinde iyi tolere edildi¤inden bevacizumab’›n bugün salvaj tedavide aktif olarak kullan›ld›¤› bildirilmektedir (23,45,74-76). Prognostik önemi bir kenara, daha intraepitelyal neoplazi evresinden itibaren serviks karsinomu için çok önemi olan ve ister glandüler isterse squamöz neoplazilerde özellikle tan›da çok de¤erli bir molekülden söz etmemek mümkün de¤il: p16INK4A, patologlar aras›nda gerek nonneoplastik ve neoplastik lezyonlar›n ay›r›m›nda, gerekse de intraepitelyal neoplazilerde yüksek riskli lezyonlar› belirleyen ve gözlemci farklar›n› ortadan kald›ran bir molekül olarak bugün rutin kullan›ma girmifl bir hücre siklus proteinidir (77,78). Gerek polarografik elektrodlarla gerekse hipoksi ile ilgili moleküller arac›l›¤› ile ölçülen tümör hipoksisi hem cerrahi olarak hem de radyoterapi ile tedavi edilen hastalarda negatif bir prognostik faktör olarak bulunmufl, multivaryant analizde de önemini korumufltur. Tümör anjiogenezisi ve anjiogenezle ilgili HIF veya karbonik anhidraz izozimleri gibi moleküllerle iliflki gösteren yay›nlar bildirilmekte ise de ise de bu konu henüz tedavide uygulamaya girmifl de¤ildir (79). Metastaz ile ilgili genlere gelirsek, kendi serimizde metastatik yolu bask›layan bir gen olan nm23’ün birçok serviks karsinomu olgusunda bulunmad›¤›, lenf nodu metastazlar› ve rekürrenslerde ise ekspresyonunun azald›¤› bulunmufltur. Ancak primer tümöre bak›larak prognoz hakk›nda öngörü yap›lamamaktad›r (57). Hücreleraras› adhezyon molekülleri aras›nda 18 J‹NEKOLOJ‹K ONKOLOJ‹ bahsedilen CD44v6 yukar›daki serimizde çal›fl›lm›fl ve multivaryant analizde ba¤›ms›z bir prognostik faktör olarak bulunmufltur (53). Literatürde DNA mismatch onar›m genleriyle serviks kanseri aras›nda hiçbir ailesel iliflki bulunamamakta ayr›ca HNPCC üyeleri ya da MMR mutayon tafl›y›c›lar›nda serviks kanseri art›m› da bildirilmemektedir. Vulva ve Vajen Kanserleri Vulva ve vajen kanserlerinde de, serviks kanserinde oldu¤u gibi prekürsör lezyonlar (VIN, VAIN) bulunmakta ve patogenezde HPV rol oynamakla birlikte serviks kanseri kadar s›k görülmemesi nedeniyle moleküler çal›flmalar ve dolay›s›yla bu konudaki bilgiler servikstekine oranla çok daha azd›r. Vulva kanseri iki farkl› yolak üzerinden oluflur: Biri, farkl›laflmam›fl-indiferansiye (warty/bazaloid) tipte VIN eflli¤inde gider, bu tipteki invaziv epidermoid karsinom komflulu¤undaki in situ karsinom alanlar›nda hemen daima HPV 16 veya 18 genomunun bulundu¤u gösterilebilmifl ancak prognostik de¤eri bildirilmemifltir. Yafll› hastalarda görülen klasik tip farkl›laflm›fl-diferansiye (simplex) keratinize epidermoid vulva-vajen kanserlerinde ise viral genom bulunmay›fl› bu iki grupta moleküler patogenezin farkl› oldu¤unu göstermektedir. Muhtemelen yüksek riskli HPV enfeksiyonu, serviks kanserlerinde oldu¤u gibi normal hücrenin tümöre dönüflmesinde haz›rlay›c› rol oynamaktad›r. Serviks karsinomlar›ndaki gibi HPV negatif vulva-vajen tümörlerinde de p53 tümör supresör gen mutasyonlar›na daha s›kl›kla rastlanmaktad›r. Anlafl›lan vulva ve vajen karsinomlar›n›n genç yafllarda görülen tipleri serviks kanserine benzer bir patogenez gösterirken, liken sklerozus birlikteli¤iyle giden ve yafll›larda görülen vulva kanserlerinde patogenez farkl›d›r (67,80). Vulva epidermoid kanserlerinde s›kl›kla saptanan 3p; 8p; 22q, inaktif X kromozomunun k›sa kolunda kay›plar olmas›, bu bölgelerde vulva kanserleri için tümör bask›lay›c› baz› genlerin bulunabilece¤ini düflündürmektedir. Kromozom 10 ve 18 uzun kollar›nda kay›p bulunan hastalarda daha agresif biyolojik davran›fl saptanmas› enterasand›r (81). Vulva kanseri hastalar›nda moleküler prognostik faktörleri de¤erlendirmeyi amaçlayan çal›flmalarda incelenen Squamous Cell Carcinoma Antigen (SCCA) saptanma yüzdesinin evre ile iliflkili olarak artt›¤› bulunmufl ancak lenf nodu metastaz› ve konvansiyonel prognostik de¤iflkenlerle istatistik iliflki bulunmam›fl olup preoperatif de¤erlendirmede yarar› gösterilememifltir (82). Birçok araflt›rmac› CD44 ekspresyonunun vulva kanserindeki önemini de¤erlendirmifltir. CD44 molekülü izoformlar› olan V4, V5, V6, V7 ve V8 ile vulva kanseri sa¤kal›m›yla iliflki gösterilememifltir, ancak evre I hastalarda V3 ve V6 ile hastal›ks›z sa¤kal›m ve genel sa¤kal›m iliflkili bulunmufltur (83). Vulva kanserinde DNA ploidisi de araflt›r›lm›flt›r. S-faz› fraksiyon analizi ile yap›lan çal›flmalarda anlaml› bir etkileflim bulunmam›flt›r. p53 ve mdm2 ekspresyonunu inceleyerek ile lenf nodu metastaz›n›n öngörülebilmesini araflt›ran bir çal›flmada iliflki gösterilememifltir. Sonuçta mevcut moleküler patolojik belirleyicilerin vulva kanserinde prognostik bak›mdan henüz pratikte uygulama konusunda yeterli olmad›¤›n› görmekteyiz (83). Endometrium Kanserleri ‹lk kez sadece klinik bulgular temelinde 1983’te Bokhman tarafindan ortaya at›lm›fl bir hipotez bafllang›c› ile, bugün için klinik, epidemiyolojik, immünhistokimyasal ve moleküler genetik özellikleri ile destek bularak, endometrial karsinom endometrioid ve nonendometrioid tipler olarak en az›ndan ikiye ayr›lmakta, morfolojik bulgular moleküler verileri desteklemektedir. Biz de bu konseptin klasik histopatoloji ve patogenetik mekanizmalar› baflar› ile integre etti¤ine inan›yoruz. Bu hipotez her ne kadar fazlaca basite indirgenmifl olsa da endometrioid karsinomun moleküler düzeyde anlafl›labilmesini sa¤lad›¤› için çok önemli olmakla birlikte, literatürdeki birçok moleküler çal›flman›n sa¤lam bir patoloji bilgisi eflli¤inde yap›lmad›¤›ndan ya da, en az›ndan histolojik tipler tam olarak ay›rdedilmeden yap›ld›¤›ndan konu detay› henüz yeterli olarak ayd›nlat›labilmifl de¤ildir. Endometrioid Tip (Tip I - Estrojen Ba¤›ml›) Endometrial Kanser Tip 1 kanserler endometrial kanserlerin %80 gibi bir oranla en çok rastlanan tipidir, ço¤u iyi farkl›laflm›flt›r ve endometrioid tip olarak isimlendirilirler, musinöz adenokanserler ve di¤er endometrioid kanser varyantlar› da reseptör içermesi ve iyi farkl›laflm›fl olmalar› nedeniyle Tip1 kanserler grubuna dahildir. Kolon karsinogenezindeki adenom-karsinom ilerleyiflinde oldu¤u gibi, endometrioid tip endometrial karsinogenezde de; gittikçe artan yap›sal ve sitolojik atipiyle beraber giden bir hiperplaziden geçerek invaziv karsinoma progresyon oldu¤una inan›lmaktad›r. Bu prekanseröz/kanser öncüsü lezyonlar için, gerek daha belirli morfolojik kriterlere dayanmas› gerekse de (belki de dolayl› olarak) gözlemcileraras› tan› farklar›n› ortadan kald›rma aç›s›ndan dünya sa¤l›k teflkilat›n›n kulland›¤› “hiperplazi” tabiri yerine “endometrial intraepitelyal neoplazi/EIN” tabirinin kullan›m›n› desteklesek ve art›k tan›da yeni tabiri kullansak da, flimdiye kadar konuyla ilgili yay›nlar›n hemen tamam›n›n klasik kullan›lagelen “hiperplazi” yi kullanmas› nedeni ile, literatürü özetlerken bu tabiri kullanmak zorunda oldu¤umuzdan; daha bafllang›çtaki hiperplazi/atipik hiperplazi/iyi farkl›laflm›fl endometrioid kanser tan›s›nda yüksek orandaki gözlemci tan› uyumsuzlu¤u nedeniyle, bu tan›lar zemininde yap›lacak moleküler çal›flmalar› de¤erlendirirken okuyucunun dikkatini yeniden çekmemiz gerekti¤ini düflünüyoruz. Endometrioid tip kanserlerde vakalar›n ço¤unda -eksojen veya endojen- karfl›lanmam›fl estrojen etkisi proliferasyon stimulusunu oluflturmaktad›r. Mevcut çal›flmalar bu tümörlerin oluflumunda PTEN tümör bask›lay›c› geni, mikrosatellit instabilitesi, PI3kinase, ras ve beta-katenin gen degiflikliklerinin en önemli de¤ifliklikler oldu¤unu, buna karfl›l›k tam aksine nonendometrioid tiplerde ise p53, her2, p16INK4A, e-kadherin de¤iflikliklerinin görülen en önemli gen de¤ifliklikleri oldu¤unu göstermifltir (fiekil 19-4). PTEN (Phosphatase &TENsin homologue): Endometrioid tip endometrial kanserlerde hem en yayg›n hem de endomet- BÖLÜM 19 ❏ Kanserin Moleküler Temeli ve Jinekolojik Kanserlerde Önemi fiEK‹L 19-4 Endometrioid (Tip I, a) ve seröz endometrial karsinogenez (Tip II, b). Bugün için bildi¤imiz genetik de¤ifliklikler ›fl›¤›nda kaba ve özet bir modeli. rioid karsinom patogenezinde erken evrede oluflan ve tümör gelifliminde merkezi rol oynayan gen kayb›d›r. Yukar›da da anlat›ld›¤› gibi, normalde PTEN proteini fosfatidil innozitol trifosfat (PIP3) defosforilasyonu sonucu AKT fosforilasyonunu engelledi¤inden, PTEN inaktivasyonu AKT fosforilasyonunu, protein sentezini ve hücre proliferasyonunu indükler. AKT aktivasyonu ayr›ca estrojen yoklu¤unda bile estrojen reseptörünün fosforilasyonuna neden olabilir, baflkaca apoptozun inhibisyonuna da neden olabilir. Atipili kompleks hiperplazi ve karsinomun ard›fl›k olarak bulundu¤u histerektomi spesmenlerinde farkl› komponentlerde idantik PTEN mutasyonlar› varl›¤›, bu mutasyonun prekürsör lezyonlardan itibaren ve invazyon geliflmeden daha önce oldu¤unu gösterir; benzer bulgular PTEN knockout farelerde de kan›tlanm›flt›r. PTEN mutasyonu atipili ve atipisiz hiperplazide %20-50 olup, endometrioid kanserde %80 oran›nda görülür, PTEN kayb› olan tümörlerin daha iyi prognozlu oldu¤una dair yay›nlar mevcuttur ancak mutasyon varl›¤› atipik hiperplazinin karsinoma ilerleyece¤ini de öngörmez (2,39). Bununla birlikte PTEN immunohistokimyas›n›n tan›da veya klonaliteyi belirlemek için kullan›m› henüz limitlidir. Cowden sendromu ve Bannayan-Zonona-Riley-Ruvalcaba sendromlar› PTEN germline mutasyonlar› sonras› oluflur, bu senderomlar›n belki de 40 yafl öncesi –erken- bafllang›çl› endometrioid endometrial kanserlerden sorumlu olabilecekleri düflünülmektedir. 19 [PIK3CA (phosphoinositide-3-kinaz)]: PTEN yola¤›nda önemli bir molekül olan PI3K nin katalitik parças› PIK3CA mutasyonlar› son y›llarda endometrioid karsinomlar›n yaklafl›k %35’inde, daha çok PTEN mutasyonu olan tümörlerde ama PTEN mutasyonu olmayanlarda da bildirilmifltir. Bu lipid kinaz molekülünün PIP24yi PIP3’e direk olarak fosforile edebilme özelli¤i, tahmin etti¤iniz gibi bir lipid fosfataz olan PTEN’in antagonisti olarak görev yapmaktad›r. Ancak PTEN mutasyonlar›n›n aksine, PIK3CA mutasyonlar›n›n komplex hiperplazilerde gözlenmemesi, bu genetik de¤isikli¤in invazyon s›ras›nda rol oynad›¤›n› ve endometrioid endometrial karsinogenezde geç bir basamak oldu¤unu düflündürmektedir (66,67,84-91). [Mikrosatellit ‹nstabilitesi (MI)]: Genomda çok yayg›n olarak bulunan polimorfik ve genellikle kodlama yapmayan bu k›sa tekrarlay›c› DNA üniteleri, yukar›da bahsedildigi gibi spesifik bir gen mutasyonundan ziyade etkin bir flekilde mismatch tamir mekanizmas› bozuklu¤unu gösterir ve sporadik tipte endometrioid endometrial kanserlerin %20’sinden ço¤unda görülür. HNPCC (hereditary non-polyposis colorectal carcinoma/LynchII sendromu) ailelerindeki endometrioid endometrial kanserlerde germline mutasyonlar sonucu ortaya ç›karken, sporadik endometrioid endometrial kanserlerde genellikle MLH1 CpG bölgesindeki promotor hipermetilasyonu sonucu inaktivasyona (=epigenetik susturma), daha az oranda da MSH2 ekspresyon kayb› ve MSH6 mutasyonuna ikincil görülürler. Klinik bak›mdan MI endometrial karsinogenede erken bir olay olup PTEN mutasyonu olan vakalarda daha s›kt›r ve daha iyi prognozla gider (2,61,87). Endometrioid kanserli bir hastada HNPCC yi flüphelendiren bulgular genç yafl, premenopozal hastal›k, alt segment yerleflim, kolon veya di¤er HNPCC tümör ve kuvvetli bir aile hikayesi varl›¤› olup bu durumda Mismatch Tamir Genlerinin genetik testi tavsiye edilmektedir (87). [KRAS]: Yap›sal aktive edici KRAS mutasyonlar›n›n hem iyi hem de az farkl›laflm›fl endometrioid karsinomlar›n %30’unda, kompleks atipik hiperplazilerin %10-15’inde görülmesi, mikrosatellit instabilitesi ile birlikte endometrioid tip endometrial karsinogeneziste klonal geliflimden önce oluflan - erken bir genetik degifliklik oldu¤unu düflündürür. KRAS mutasyonlar›n›n ço¤u ekson 1 (kodon 12-13) ve ekson 2 (kodon 61) yerleflimli olup metilasyonla iliflkili GC-AT transizyonlar›yla karakterlidir. Ras onkogen ekspresyonu ile kliniko-patolojik prognostik de¤iflkenler aras›nda bir iliflki bulunmam›flt›r. β-katenin (CTNNB1)]: E-kadherin ile kompleks yaparak hücre adhezyonu ve invazyonda rol oynayan, ayn› zamanda wnt sinyal yola¤›n›n önemli bir parças› olan bu molekülün aktive edici mutasyonlar›na endometrioid adenokarsinomlar›n %25-44’ünde rastlanmakla beraber, neoplastik geliflimdeki fonksiyonu tam olarak anlafl›lamam›flt›r. Ekson 3 mutasyonlar› protein stabilizasyonu ve nükleer protein birikimi ile transkripsiyon aktivasyonuna neden olur. APC proteini de β-katenin düzeylerini azaltmaktad›r. β-katenin mutasyonlar›n›n PTEN, MI veya RAS gibi di¤er genlerle iliflki göstermemesi, bu genin özellikle skuamöz diferensiasyon gösteren Tip I endo- 20 J‹NEKOLOJ‹K ONKOLOJ‹ TABLO 19-6 Tip I ve Tip II Endometrial Kanserlerin Bugün Bilinen Klinik ve Moleküler Özellikleri Özellik Yafl Klinik Morfoloji Prekürsor Non-neoplastik Endometrium Moleküler Genetik Klinik Gidifl Yay›l›m Tip I 55-65y Karfl›lanmam›fl estrojen Obesite Hipertansiyon Diabet Endometrioid Atipik hiperplazi (EIN) Hiperplastik PTEN inaktivasyonu MI KRAS β-katenin PIK3CA Di¤er Hissettirmeden, yavafl Lenfatiklerle metrial karsinogeneziste ba¤›ms›z bir rolü oldu¤unu düflündürmektedir. Premalign lezyonlarda varl›¤› nedeniyle karsinogenezde erken safhada rolü oldu¤u, ama daha sonra da progresyonda da rol oynad›¤› düflünülmektedir. Di¤er taraftan hücreleraras› ba¤lant› molekülü olan E-kadherin ekspresyonu azalmas›, hiperplaziler ve tip 1 endometrial kanserlerde görülmez. p53 tümör bask›lay›c› gen mutasyonlar› di¤er tümörlerde oldu¤u gibi endometrioid tip endometrial kanserlerde de çal›fl›lm›flt›r. Bugün için, endometrioid tip endometrium kanserlerinde p53 mutasyonlar›n›n az farkl›laflm›fl (grade 3) ve ileri evre tümörlerde görüldü¤ü, bu nedenle p53 geninin endometrioid tip endometrial kanserlerde inisiasyonda de¤il progresyonda rol oynad›¤› anlafl›lm›flt›r (25,29,86). Prognostik anlamda k›saca MI veya PTEN ve beta katenin(CTNNB-1) varl›¤› tip I kanserler için iyi prognozu, p53 ve PIK3CA mutasyonlar› varl›¤› ise aggressif davran›fl› gösterir. Endometrioid tümörlerde PI3K/AKT/MTOR ve proteozom inhibitorleri gibi apoptoz rezistans genlerini hedefleyen “hedefe yönelik moleküler tedaviler” ilerde umut verici olabilir. Non-Endometrioid Tip (Tip II -Estrojen Ba¤›ml› Olmayan) Endometrial Kanser: Daha nadir olan, sporadik endometrial karsinomlar›n %10-20 sini oluflturan non-endometrioid tümörler hakk›nda bilgilerimiz daha s›n›rl›d›r. Tan›m olarak az farkl›laflm›fl (grade 3) olup tip I kanserlerin aksine atrofik endometrium zemininde geliflirler. Non-endometrioid tip kanserlerin prototipi, %10 oranda görülen, overin seröz kanserleri ile olan morfolojik ve biyolojik benzerlikleri nedeni ile aynen isimlendirilen seröz karsinomdur (uterin seröz papiller kanser). Prekürsör lezyon endometrial glandüler displazi (EmGD) olarak isimlendirilir. p53 gen anomalilerinin %100’e yak›n oranda bulunuflu, intraepitelyal kanserlerde bile çok yüksek oranda olmas› özellikle dikkat çe- Tip II 65-75y Atrofi Zay›f/normal Seröz (CCCa-Karsinosarkom) EGD/EICa Atrofik p53 Her2/Neu P16INK4A E-kadherin Genetik ‹nstabilite Di¤er Agresiv IP ve lenfatiklerle kici olup p16, c-ERBB2, E-kadherin mutasyonlar›, mitotik a¤ protein de¤ifliklikleri ve kromozom instabilitesi ile karakterlidirler. Bu bulgular, PTEN, K-ras ve MI olmay›fl› ile birlikte endometrioid tümörler ile USPC aras›nda önemli bir fark oluflturur. Endometrioid ve non-endometrioid kanserler aras›nda bugün için bilinen moleküler farkl›l›klar Tablo 19-6’da özetlenerek sunulmaktad›r. fieffaf hücreli kanserler ve MMMT patogenezi hakk›nda bilinenler daha azd›r. Uterin Seröz Papiller Kanser (USPC) ve Prekürsörlerindeki Gen De¤ifliklikleri (fiekil 19-4) [p53]: Seröz endometrial karsinogenezin inisiasyon ve progresyonu tam olarak anlafl›lamam›fl ise de, en belirgin ve en s›k görülen moleküler degifliklik anormal protein birikimi sonucu immunhistokimyasal olarak kuvvetli ve diffüz nükleer boyanmaya neden olan p53 tümör bask›lay›c› gen missens mutasyonlar›d›r. Germline p53 mutasyonlar› ve kansere afl›r› predispozisyonla karakterli Li-Fraumeni ailelerinde uterin seröz kanser olmamas›, uterin seröz kanser patogenezinde p53 mutasyonu d›fl›nda baflka genetik de¤iflikliklerin de sorumlu oldu¤unu gösterir. Endometrial intraepitelial karsinom (EIC), seröz karsinoma eflde¤er hücrelerden oluflmakla birlikte stromal invazyon göstermez; p53 mutasyonlar› yaklafl›k %75 orandad›r, bu nedenle EIC seröz kanserin bir basama¤› olarak belirir. Fadare ve Zheng, EIC’nin premalign lezyon de¤il, intraepitelyal da olsa malign bir neoplaziyi yans›tmas› gerekçesi ile morfolojik olarak tan›nabilen ilk gerçek prekürsörün endometrial glandüler displazi (EmGD) oldu¤unu, EIC tan›m›n›n bir morfolojik tarif oldu¤unu ama bir tan› olmad›¤›n› bildirmektedir. EIC, ço¤u vakada ekstrauterin hastal›kla birlikte bulundu¤undan asl›nda bir endometrial seröz karsinomdur ve karsinom gibi tedavi edilmelidir. Morfolojik olarak “normal” ama p53 immünreaktif hücreler “p53 signatures” olarak isim al›r ve progresyon modelinde bafllang›çtan itibaren seröz karsi- BÖLÜM 19 ❏ Kanserin Moleküler Temeli ve Jinekolojik Kanserlerde Önemi nomlarla birliktelik gösterir; bu nedenle immunhistokimyasal olarak en erken tan›nabilen prekürsör lezyon olma özelli¤ini tafl›r; seröz kanserlerin %50’sinden ço¤unda bulunur, gros olarak görülemez, atipi içerir ama anaplazi içermez, beraberinde EIC foküseri ve geçifl alanlar› içerir. Mikrodisseksiyonla bu alanlarda p53 allel kayb› ile di¤er mikrosatellit polimorfik DNA de¤ifliklikleri saptan›r. [HER2/neu]: Seröz endometrial kanserlerde %45 overekspresyonu ve %70 gen amplifikasyonu saptanan bu molekül, ayni zamanda az farkl›laflm›fl (grade 3) endometrioid tip ve mikst tip endometrial adenokanserde de anormallik gösterir. Seröz kanserlerde immünhistokimyasal 2+/3+ HER2/neu overekspresyonu yaklafl›k %32 oranda olup bu vakalar›n yar›s›ndan ço¤unda FISH ile amplifikasyon saptan›r. Bu bulgular HER2/neu overekspresyonunun p53 mutasyonlar› ile beraber, hem seröz tip endometrial kanserlerde, hem de endometrioid tip endometrial adenokarsinomun dedifferensiasyonunda önemli rolü oldu¤unu gösterir. [P16INK4A]: Seröz kanserlerin %92’sinden fazlas›nda diffüz ve kuvvetli, -arada negatif hücre atlay›fl› olmayan- bir P16INK4A immunhistokimyasal pozitivitesi saptan›r. Bu bulgu, seröz tip endometrial kanserde hem p16INK4A/cyclinDCDK/pRb-E2F, hem de ARF-MDM2-p53 hücre siklusu yolaklar›n›n ifle kar›flt›¤›n› gösterir. P16 ekspresyonu hem p53 gen mutasyonu ile koreledir hem de genifl serilerde agresif tumör davran›fl›n› belirler. Biyopsi materyellerinde adenokarsinomun endosevikal ya da endometrial orijinli oldu¤unun ay›rdedilmesinde IHC kullan›l›rken seröz tip endometrial kanserdeki P16INK4A pozitivitesi asla ak›ldan ç›kar›lmamal›d›r. [E-kadherin]: Sitoplazmada katenin ile kompleks yaparak aktin iskeletine tutunan bu molekülün ekspresyon azl›¤› hücre kohezyonunun kayb› ile birlikte gider. Non-endometrioid (seröz ve fleffaf hücreli) tümörlerde %60 tamamen, %87 oranda da k›smi olarak E-kadherin ekspresyon kayb› vard›r ve kötü prognozun habercisidir. Daha az oranda az farkl›laflm›fl endometrioid tümörlerde de bildirilmifltir. [Kromozom instabilitesi (Genetik instabilite)]: Birçok kromozomda ama özellikle kromozom 1p32-33 bölgesinde heterozigosite kayb› (LOH) seröz karsinomlar›n tipik bulgusudur, %60’in üzerinde bir oranda saptan›r ve anöploidi varl›¤›n›n bir yans›mas›d›r (Bu antitenin MI ile farkl› bir antite oldu¤una tekrar dikkat çekmemiz gerekir). Mikst Endometrial Karsinomlar Yukar›daki bulgulardan, tip I kanserlerle seröz kanserler aras›nda moleküler de¤iflikliklerin farkl›l›¤› aç›kça görülmektedir. Ancak seröz endometrial karsinomlar›n az da olsa bir k›sm›, tip II moleküler de¤ifliklikler yan›s›ra, ayn› tümörde, tümörün farkl› bölgelerinde endometrioid (tip I) endometrial karsinoma protein ekspresyon paterni göstermektedir. Bu bulgu, tip II endometrial kanserlerin en az›ndan bir k›sm›n›n, seröz kansere öncelik eden endometrioid tipte endometrial kanserin varl›¤›n›; ayr›ca, seröz kanserin onun zemininde ve dedifferansiasyonu ile olufltu¤unu düflündürmektedir. Bu hipotez ayn› za- 21 manda mikst tip (tip I + tip II) endometrial karsinomlar› da aç›kça izah eder (89-91). Son olarak Malign Mikst Müllerien Tümörler (MMMT) konusunda bir özet yapacak olursak, art›k bugünkü bilgilerimiz ›fl›¤›nda MMMT oluflumunda epitelyal (karsinomatöz) komponent ile mezenkimal (sarkomatöz) komponentin ayn› pluripotent kök hücreden köken ald›¤›n› düflünen “kombinasyon” teorisi ile, sarkomatöz komponentin karsinomatöz komponentin dediferansiasyonu ile geliflti¤ini öne süren “konversiyon” teorilerinin kabul gördü¤ünü söylemeliyiz. Moleküler veriler (Xkromozom inaktivasyonu, LOH ve p53 mutasyon paterni ile elde edilen sonuçlar), her iki komponentin ayn› klondan geliflti¤ini göstermektedir. Doku kültürlerinde de, karsinom komponenti sarkoma dönüflebilmekte, bifazik morfolojik ve immünhistokimyasal özellikler ortaya ç›kabilmektedir (66,67). OVER KANSERLER‹ Jinekolojik maligniteler aras›nda en ölümcülü olan over kanserlerinin tan› ve tedavisindeki güçlük, bu agresif tümörlerin patogenezini anlamakta da mevcuttur. Bu güçlük temelinde, histolojik ve histogenetik farkl›l›klara ra¤men uzun y›llar over kanserlerinde yap›lan çal›flmalar›n sanki tek bir hastal›k imifl gibi çeflitli histolojik subtiplerinin hatta epitelyal-stromal ve/veya germ hücreli tümörlerinin ay›rdedilmeden, yani histopatoloji temeli olmadan yürütülmüfl olmas› yan›s›ra hastal›¤›n genellikle geç evrede tesbit edilebilmesi nedeniyle erken evre kanserlere ve premalign lezyonlara ait örnekleme yap›labilmesinin de imkans›zl›¤›, serviksteki servikal intraepitelyal lezyonlar (SIL) ve endometriumdaki hiperplaziler gibi overin kansere dönüflen prekürsör lezyonlar› hakk›nda hem fikir hem de tabir birli¤ine var›lamam›fl olmas› gibi etkenler yatar. Histolojik olarak malign bir tümörde benign alanlar›n veya transizyon bölgelerinin bulunmas›, malign tümörün benign tümör zemininde geliflti¤ini göstermek için yeterli olamamaktad›r. Moleküler genetik teknikler yard›m›yla klonal büyümenin benign ya da düflük malignite potansiyelli tümördeki hücre ile ayn› moleküler de¤ifliklikleri paylaflt›¤›n› göstererek, etyopatogenezdeki ortakl›¤› a盤a ç›karmak en do¤ru yol diyebiliriz. Over tümörlerinde tan›mlanan bu güçlüklere ra¤men, over tümörigenezisinde en az›ndan baz› moleküler olaylar›n a盤a ç›kar›lmas›nda flu güncel ve önemli geliflmeler inkar edilemez: Over kanserlerinin ço¤u Müller kanal›ndan geliflen hücrelere benzer, ama bilindi¤i gibi overde müller orijinli hücre yoktur. Bu nedenle geçmiflte, over tümörlerinin over yüzey epitelinden geliflti¤i, seröz-musinöz-endometrioid ve fleffaf hücreli tümörlerinin ayn› hücreden köken ald›¤› hatta borderline ve malign over tümörlerinin tamamen ard›fl›k basamaklar oldu¤u düflünülmüfl ve s›n›flamalar buna göre yap›lm›fl idi. Bugün için (s›n›flamalar henüz de¤iflmese de) yüksek grade’li seröz kanserlerin ço¤unun fallopian tüplerin fimbrial ucundaki epitelden köken ald›¤›, fleffaf hücreli ve endometrioid karsinom öncüsünün de endometriozis oldu¤unu düflündüren bulgular yo¤unluk kazanm›flt›r. Bu bulgular bügünkü over kanseri orijini ile ilgili bilgilerimizdeki en büyük de¤ifliklikleri 22 J‹NEKOLOJ‹K ONKOLOJ‹ oluflturmakla kalmamakta, önlem ve tedavi stratejilerinde de 盤›r açacaklar› düflünülmektedir: Rutin histerektomi s›ras›nda tubalar›n da ç›kar›lmas›, ya da tüp ligasyonu yerine salpinjektomi, ilerde yüksek grade’li seröz kanserlerin oluflumunu engelleyecek, bu hastalarda in vitro fertilization sayesinde fertilite de korunabilecektir. fieffaf hücreli ve endometrioid kanserler ise endometriozis tedavisindeki geliflmeler sayesinde engellenebilecektir. fieffaf hücreli ve endometrioid kanserlerde, afla¤›da anlat›lacak ARID1A proteini ekspresyon kayb› hem over hem de uterus orijinli tümörlerde birkaç ay önce bildirilmifltir. Kökenlerinin ayn› -endometrial epitel- oldu¤u göz önüne al›n›rsa bu durum hiç de flafl›rt›c› de¤ildir ve tümörun köken ald›¤› organa bak›lmaks›z›n tedavide de benzer uygulamalar önerilebilece¤ini düflündürür. Bu yaklafl›m, klinik araflt›rmalarda birbirleri ile hiç iliflkisi olmayan seröz kanserlerle fleffaf hücreli ve endometrioid tümörlerin birlikte de¤erlendirilmesi yerine; histolojik, moleküler ve embriyolojik benzerlikleri olan uterus ve over fleffaf hücreli/endometrioid kanserlerinin birarada de¤erlendirilmesinin çok daha mant›kl› oldu¤u görüflünü de düflündürür ve destekler. Yeni bir Over Kanseri Patogenezi Modeli Over kanserinin overden bafllay›p sistematik olarak pelvise ve abdomene yay›ld›¤›, bafllang›çtaki iyi farkl›laflm›fl tümörün zamanla az farkl›laflmaya u¤rad›¤›n› varsayan modelin klinik gerçeklere ve moleküler bulgulara uymamas› uzerine Shih ve Kurman taraf›ndan öne sürülmüfl olan yeni bir over kanser patogenezi modelini özetleyece¤iz (Tablo 19-7) (2,7,50,67,92-101): Bu model, daha önce overin yüzey epitelinden geliflti¤ini düflündü¤ümüz tümörleri, tümör oluflum modeli ve karakteristik moleküler de¤ifliklikleri göz önüne alarak, endometrium TABLO 19-7 kanserinde oldu¤u gibi -ama overde histopatolojik ve diagnostik temeli olmaks›z›n- Tip I ve Tip II kanserler olmak üzere 2 genel kategoriye ay›r›r. Yazarlar, özellikle daha az görülen tiplerde ve morfolojik olarak çak›flan kategorilerde moleküler de¤ifliklikler hakk›nda bilgilerimiz artt›kça bu klasifikasyon sisteminin daha da geliflece¤ini düflündüklerini söylemektedirler (66,67,93,94). K›saca özetleyecek olursak Tip I tümörler genellikle düflük grade’li tümörler olup yavafl seyirli ve nisbeten iyi davranan, klinik olarak lokalize iken saptanan, erken evrede tesbit edilen tümör grubunu oluflturmaktad›r, genetik instabilite göstermezler, yine genellikle borderline tümörler zemininde (serözler seröz borderline tümör; müsinözler müsinöz borderline tümörler; endometrioidler ve fleffaf hücreliler ise endometriotik kist zemininde) geliflir, genetik profilde p53 mutasyonlar› gözlenmez, p53 d›fl›ndaki genlerde de¤ifliklik vard›r: seröz ve müsinoz tümörler KRAS/BRAF mutasyonlar›, endometrioid tümörler ise tipI (endometrioid tip) endometrioid kanserlerde oldu¤u gibi beta-katenin/PI3K/PTEN/ARID1 gen de¤ifliklikliklerini içermektedir (fiekil 19-5). Yüksek gradeli Tip II tümörlerde ise (yüksek gradeli seröz, yüksek gradeli endometrioid, indiferansiye ve MMMT) ortak karakter olarak p53 mutasyonu ve BRCA1/2 kayb› ile giden genetik instabilite söz konusudur (fiekil 19-6). Yüksek grade’li tümörler, primer over tümörlerinin bat› dünyas›nda en s›k görülen grubunu oluflturur ve hastalar›n %90’dan fazlas› ileri evre invaziv tümörler olarak baflvurur, klinik olarak da agresiv davran›rlar. Her ne kadar farkl› histomorfolojik tiplerde farkl› genotipler görülüyor olsa ve moleküler yolaklar farkl› olsa da Tip I ve Tip II tümörlerin orijinlerinde bafllang›ç olay genellikle ortak gibi görünmektedir (66,67,90,93). Düflük grade’li (Tip I) Tip I ve Tip II Over Kanserlerinin Karfl›laflt›rmal› Özellikleri (7,50,67,92-102) Prekürsör Muhtemel Orijin En S›k Mutasyon Kromozom ‹nstabilitesi Makroskopi Tip I Tümörler Düflük Grade Seröz Ca MIC Düflük Grade Endometrioid Endometriosis, MIC Ca Endometriosis, MIC fieffaf Hücreli Ca Müsinöz Ca (intestinal) MIC Müsinöz Ca (müllerian) Endometriosis,MIC Tip II Tümörler Endosalpinks, Yüksek Grade Seroz Ca endosalpingiozis (over/fimbria/periton) Yüksek Grade Endometrioid Endometriosis, MIC Ca Endosalpinks, Indiferansiye Ca endosalpingiozis, Endometriosis, MIC KA, APST, NI-MPSC KRAS, BRAF Endometrioma/emBL HOXA10,CTNNB1, Az PTEN, ARID1 Az PIK3CA, ARID1 Az Bilateral, intrakistik/ peritoneal yüzey yayg›n Uni/bilateral, lokalize HOXA11, KRAS, BRAF, c-myc KRAS Az Unilateral, lokalize, uzak yay›l›m Unilateral,lokalize Az Uni/bilateral,lokalize p53 signature, TICa p53 Endometrioid p53 ? p53 Çok Dominant kitle, bilateral, peritoneal yay›l›m Çok Dominant kitle, Uni/ bilateral Dominant kitle, Uni/ ? bilateral, peritoneal yay›l›m Endometriosis/ Endometrioma MIC, APMT MIC,Endometriosis/ Endometrioma MIC; Müller inklüzyon kisti, KA; kistadenoma, APST; atipik proliferatif seröz tümör, NI-MPSC; noninvaziv mikropapiller seröz kanser, EmBL; endometrioid borderline, APMT; atpik proliferatif musinöz tümör, TICa; tubal intraepitelyal kanser BÖLÜM 19 ❏ Kanserin Moleküler Temeli ve Jinekolojik Kanserlerde Önemi 23 (A) fiEK‹L 19-5 (A) Düflük grade`li (Tip I) ve (B) Yüksek Grade`li (Tip II) over karsinogenezi hipotetik modeline ait bugün bildi¤imiz genetik de¤iflikliklerin kabaca özeti. (B) kanserlerde öncelikle homeobox (HOX) genlerinden HOX10 neoplastik transformasyon öncesi de¤iflik epitelyal farkl›laflmay› belirleyecek flekilde bir anlamda metaplastik bir de¤iflikli¤e u¤rar. Sonraki basamaklarda yukarda bahsedilen ve Tablo 19-7’de özetlenen di¤er genler ifle kar›fl›r. Yüksek grade’li (Tip II) kanserlerin oluflumunda ise ortak epitelin tuba uterina epiteli oldu¤una dair deliller öncelikle BRCA1/2 ailelerinde yap›lan çal›flmalarla ortaya ç›kar›lm›fl olup gün geçtikçe bu hipotezin kabul görmesini destekleyen deliller de artm›fl ve artmaktad›r. Over kanseri konusunda yap›lan çal›flmalar›n sadece en yo¤un, güncel ve önemli olmakla kalmay›p over karsinogenezi hakk›ndaki bilgilerimize katk›s› inkar edilemeyecek kadar çok olanlar› hiç kuflkusuz herediter over kanseri ile ilgili olanlard›r. BRCA 1’in genetik kayb› ailesel over kanseri içinde çok önemli bir genetik de¤iflikliktir. BRCA 1’de germline mutasyonlar› tafl›yan kad›nlar over kanserine daha genç yaflta (yaklafl›k on y›l erken) yakalan›r. BRCA 1 tafl›y›c› hastalarda ileri evre over kanserinde bile önemli bir sa¤kal›m avantaj›na yol açan ve hastal›¤› ›l›ml› olarak nitelemeye yol açan bir fark bulunmufltur. BRCA 1 ve 2 nin prognostik önemi de henüz tart›flmal› konulardand›r. Ailevi BRCA1 gen mutasyonu tafl›yan hastalarda geliflen over kanserleri hemen daima seröz papiller niteliktedir. Herediter over kanseri ile ilgili BRCA1/2 ailelerinde yap›lan çal›flmalarla son y›llarda ortaya ç›kar›lm›fl gerçekten çok önemli ve flafl›rt›c› bir geliflme ise yüksek grade’li (Tip II) over kanserlerinin oluflumunda köken hücrenin tüba uterina epiteli oldu¤udur (91,93,94,103,104). 24 J‹NEKOLOJ‹K ONKOLOJ‹ Yüksek grade’li seröz karsinogenezis ilerleyifli basamak detay›yla ilgili son bilgiler Norquist taraf›ndan yap›lan çok ak›ll›ca bir çal›flmayla ortaya at›lm›fl olup flu flekilde özetlenebilir (fiekil 5; 103,104): BRCA1 mutasyonu olan kiflilerde BRCA1 genlerinden birinin mutant di¤erinin fonksiyonel olmas›, di¤er bir isimle haploinsufficiency, ilk etapta tuba uterina distal ucu epitel hücrelerinde Ki-67 ve di¤er proliferasyon belirleyicileri ile gözlenebilecek bir ço¤almaya, ço¤alan hücrelerde bu süreçte p53 mutasyonu ile fonksiyon kayb› olmas› morfolojik olarak normal ama immünhistokimyasal olarak p53 anormal proteini birikimi ile karakterli ortak prekürsör -p53 signatürlerinin- ortaya ç›kmas›na neden olur (ard›fl›k p53 nukleus pozitivitesi ile karakterli 10-12 epitel hücresi). p27 hücre siklus proteini (cdki) kayb› genellikle p53 kayb›yla eflzamanl›d›r. Takibeden genetik basamak olarak BRCA1 allelerinden ikincisinin kayb› neoplastik proliferasyonu belirler. E¤er p53 mutasyonu olmasa BRCA allellerinden ikisinin de kayb›n›n, biriken DNA hasar›n›n tamiri için hücre büyümesini durdurmas› gerekir iken; p53 mutasyonu varl›¤› nedeni ile DNA hasarl› hücreler ço¤almaya devam eder ve genomik instabilite artar. Bu basamakta hücreler hala intraepitelyal olmakla birlikte morfolojik olarak malignite kriterleri tafl›makta olup seröz tübal intraepitelyal neoplazi (ya da sadece tübal intraepitelyal neoplazi, baz› yazarlarca da (seröz) tübal intraepitelyal karsinoma (STIC,TIC) ismiyle tan›mlan›r. Bu neoplastik hücreler kolayca peritona, over yüzeyine, over stromas›na ya da tüp lümeninde uygun bulduklar› pek çok yere yerleflebilir ve ço¤al›r. Bu çal›flma, p53 geni allelerinden birinde kay›pla giden (p53 geni bak›m›ndan haploinsufficiency gösteren) Li-Fraumeni ailelerinde neden yüksek grade’li seröz kanserlere s›k rastlanmad›¤› sorusuna da cevap getirmifltir. p53 mutasyonlar› yüksek grade’li seröz kanserlerin nerede ise “olmazsa olmaz›” olmas›na ra¤men, allellerden birinde ailevi olarak mutasyon nedeni ile sadece bir tane fonksiyonel allel bulunan, bu nedenle ikinci allelin daha çabuk ve kolay kaybolaca¤› düflünülen Li Fraumeni ailelerinde yukar›da bahsedildigi gibi pek çok kansere rastland›¤› halde uterin seröz kanserler veya tuba uterina-over ya da peritonun yüksek grade’li seröz kanserlerinin görülmemesinin nedeni, muhemelen Li-Fraumeni ailelerinde BRCA1 ve BRCA2 genlerinin intakt olmas›d›r. Bu bulgu ayr›ca p53 ve BRCA gen kay›plar›n›n seröz karsinogenezin erken döneminde inkar edilmez derecede önemli bir ikili oldu¤unu da ispatlar. Yukarda anlat›lan bilgileri, al›flageldigimiz tabirle “yüzey epitelinden geliflen tümörlerle” k›smen ba¤daflt›rarak bir özet yapmam›z gerekirse, art›k “seröz” over kanserinin düflük ve yüksek grade`li seröz kanserler olmak üzere iki farkl› ana gruptan olufltu¤unu biliyoruz: Düflük grade`li (iyi farkl›laflm›fl) seröz kanser seyrek olup moleküler olarak seröz borderline tümörlere benzemekte, nadir de olsa baz›lar› histopatolojik birliktelik de göstermekte, KRAS ve BRAF onkogen de¤ifliklikleri en önemli moleküler de¤ifliklikleri oluflturmaktad›r. Bu grup tümörlerde p53 mutasyonlar› yok denecek kadar nadirdir. Yüksek grade`li (orta ve az farkl›laflm›fl) seröz kanser ise aksine çok yüksek oranda p53 mutasyonu içerir, KRAS ve BRAF mutasyonu içermez; bafllang›çta BRCA1-BRCA2 ailelerindeki yüksek grade`li seröz kanserlerde saptand›¤› gibi tuba uterina fimbrial ucundaki epitelden köken al›r. Primer peritoneal kanserler de bu gruptad›r ve moleküler, klinik ve tedavi bak›m›ndan eflde¤er oldugu düflünülmektedir. Art›k seröz over kanserlerinin histopatolojik gradelemesinde ikili (binary) grade sistemi kullan›lmas›n›n bügünkü moleküler bilgilere ve karsinogenez modeline daha iyi uymakla kalmay›p, patologlararas› tan› uyumu sa¤lama aç›s›ndan da tercih edildigini söylememiz gerekir (100-102). Müsinöz kanserlerde unutulmamas› gereken konu, gerçek primer müsinöz kanserlerin oldukça nadir oldu¤u, histopatolojik olarak metastatik (ço¤u apendiks, pankreas, di¤er GI) kanserler ekarte edilmeden tedaviye bafllanmas›n›n uygun olmad›¤›d›r. Bu nadir tümörler hem histopatolojik hem de moleküler anlamda belirgin bir benign-borderline intraepitleyal kanser ve invaziv malign tümör geçiflleri gosterir. Moleküler olarak da histolojik progresyonla orant›l› olarak artan kras mutasyonu söz konusudur. Bu bilgilerden anlafl›labilece¤i üzere seröz tümörlerden farkl› olarak, benign ya da borderline musinöz tümörlerin opere edilmesinin ileride geliflecek bir müsinöz karsinomu engelleyece¤ini varsaymak yerinde bir görüfltür. Endometrioid ve fleffaf hücreli over kanserleri, uterusun endometrioid tipteki (tip I) kanserleriyle hem histolojik hem de moleküler benzerlikler gösterirler, özetle PTEN tümör bask›lay›c› geni, mikrosatellit instabilitesi, ras ve beta-katenin gen degifliklikleri ile k›sa bir süre bildirilen kromatin biçimlenmesinde rol oynayan ARID1 geni anormalliklerinin en önemli de¤ifliklikler oldu¤unu söylememiz gerekir. Endometrial kanserlerin orijininin endometrium, endometrioid ve fleffaf hücreli over kanserlerin orijininin de endometriozis (ektopik endometrium) oldu¤unu düflünürsek bunu bir benzerlikten ziyade “eflde¤erlik” olarak yorumlamam›z daha do¤ru olur (99, 105,106). Az farkl›laflm›fl endometrioid ve MMMT’nin ise yüksek grade’li seröz kanserlere eflde¤er oldu¤u unutulmamald›r. KAYNAKLAR 1. 2. 3. 4. 5. 6. 7. 8. 9. Knudson AG. Cancer genetics. Am J Med Genet 111:96, 2002 Neoplasia. Robbins and Cotran. Pathologic Basis of Disesae, 8th edition. Edited by Kumar, Abbas, Fausto Aster. Sounders, 2010 The Genetic Basis of Human Cancer, 2nd ed. Vogelstein B, Kinzler KW (eds) (2002). New York: McGraw-Hill. Narod S. Modifiers of risk of hereditary breast cancer. Oncogene 25:5832, 2005 Rustgi A. The genetics of hereditary colon cancer. Genes Dev 21:2525, 2007 Weinberg RA, Halahan D. The hallmarks of cancer. Cell 100:57, 2000 Khalique LR, Ayhan A, Weale ME, Jacobs IJ, Ramus SJ, Gayther SA. Genetic intra-tumour heterogeneity in epithelial ovarian cancer and its implications for molecular diagnosis of tumours. J Pathology 211(3):286-295, 2007 Ayhan A, Söylemezo¤lu F. Tümör hücre kineti¤i. Ankara Patoloji Derne¤i Bülteni. 13:5-8. 1996 http://www.nature. com /ncb /celldivision/ BÖLÜM 19 ❏ Kanserin Moleküler Temeli ve Jinekolojik Kanserlerde Önemi 10. Kajikawa K, Yasui W, Ayhan A, ve ark. Expression of Epidermal Growth Factor in human tissues. Virchows Arch A Pathol Anat 418:27-32, 1991 11. Kameda T, Yasui W, Tsujino H, Ayhan A, Tahara E. Tyrosine kinase activity of epidermal growth factor receptor in human gastric carcinomas. Path Res Pract 188:37-43, 1992 12. Kitadai Y, Yasui W, Yokozaki H, Ayhan A ve ark. Expression of amphiregulin, a novel gene of the epidermal growth factor family, in human gastric carcinomas. Jpn J Cancer Res 84:879-884, 1993 13. Ertoy D, Ayhan A, Saraç E, ve ark.: Clinicopathological implication of cripto expression in early stage invasive cervical carcinomas. Eur J Cancer 36 (8):1002-1007, 2000 14. Baykal C, Ayhan A, Al A, Yüve K, Ayhan A. Overexpression of the c-Met/HGF receptor and its prognostic significance in uterine cervix carcinomas. Gynecol Oncol 88(2):123-9, 2003 15. Baykal C, Al A, Ayhan A ve ark. Comparison of HGF levels of epithelial ovarian cancer cyst fluids with benign ovarian cysts. Int J Gynecol Cancer 13:771-775, 2003 16. Gtschwind A, Fischer O and Ullrich A. The discovery of receptor thyrosine kinases: targets for cancer therapy. Nature Reviews Cancer 4:361-70, 2004 17. Ayhan A, Ertunç D, Tok EC, Ayhan A. Expression of the c-met in advanced epithelial ovarian cancer and its prognostic significance. Int J Gynecol Cancer 15(4):618-23, 2005 18. Kulbe H, Thompson R, Wilson J, Robinson S, Hagemann T, Fatah R, Gould D, Ayhan A, Balkwill F. The inflammatory cytokine TNF-a generates an autocrine tumor-promoting network in epithelial ovarian cancer cells. Cancer Research 67(2)585-592, 2007 19. Collard JG. Kip moving. Nature 428: 705-707. 2004 20. De Vita, Hellman and Rosenberg’s Cancer,-Principles and practice of oncology. Editors : De Vita VT, Lawrence TS, Rosenberg, Walters Kluwer-Lipponcott Williams & Wilkins, 2008 21. Fearon ER and Vogelstein BA. A genetic model for colorectal tumorigenesis. Cell 61:759-767, 1990 22. Yasui W, Ayhan A, Kitadai Y, ve ark. Increased expression pf p34cdc2 and its kinase activity in human gastric and colonic carcinomas. Int J Cancer 53:36-41, 1993 23. http://www.hopkins-coloncancer.org/subspecialities/ 24. ttp://pathology.jhu.edu/pancreas.panin 25. Ayhan A. Gündemdeki Molekül: p53. Hacettepe T›p Dergisi 28: 84-9, 1997 26. Palmero EI, Achatz MI, Ashton-Prolla P, Olivier M, Hainaut P. Tumor protein 53 mutations and inherited cancer: beyond LiFraumeni syndrome. Curr Opin Oncol 22(1):64-9, 2010 27. Ayhan A, Yasui W, Yokozaki H, ve ark. Genetic abnormalities and expression of p53 in human colon carcinomas. Int J Oncol 1:431437, 1992 28. Yokozaki H, Kuniyasu H, Ayhan A, ve ark. p53 point mutations in primary human gastric carcinomas. J Cancer Res Clin Oncol 119:67-70, 1992 29. Ayhan A., Tuncer S, Ayhan A., ve ark. Abnormal expression of cripto and p53 protein in endometrial carcinoma and its precursor lesions. Eur J Gynecol Oncol 19(3):316-319, 1998 30. Ayhan A, Tuncer ZS, Ayhan A. p53 expression in serous ovarian carcinoma with regard to second look findings. Eur J Gynecol Oncol 19(5):501-503, 1998 31. He L, He X, Lowe SW, Hannon GJ. microRNAs join the p53 network—another piece in the tumour-suppression puzzle. Nat Rev Cancer 7(11):819-22, 2007 32. Shmueli A, Oren M. Mdm2: p53’s lifesaver? Mol Cell 23;25(6):794-6, 2007 33. Khoury MP, Bourdon JC. The isoforms of the p53 protein. Cold Spring Harb Perspect Biol 2(3):a000927, 2010 25 34. Lee WH and Boyer TG. BRCA1 and BRCA2 in breast cancer. The Lancet supplement. 358:5-10, 2001 35. Zheng L, Li S, Boyer TG. Lessons learned from BRCA1 and BRCA2. Oncogene 6159-75, 2001 36. Ramus SJ, Pharoah P, Ayhan A, ve ark. BRCA1/2 mutation status influences somatic genet›c progression in inherited and sporadic epithelial ovarian cancer cases. Cancer Res 63:417-423, 2003 37. Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATRChk1 pathways in DNA damage signaling and cancer. Adv Cancer Res 108:73-112, 2010 38. Tischkowitz M, Xia B. PALB2/FANCN: recombining cancer and Fanconi anemia. Cancer Res 70(19):7353-9, 2010 39. Jiang BH, Liu LZ. PI3K/PTEN signaling in angiogenesis and tumorigenesis. Adv Cancer Res 102:19-65, 2009 40. Markman B, Atzori F, Pérez-García J, Tabernero J, Baselga J. Status of PI3K inhibition and biomarker development in cancer therapeutics. Ann Oncol 21(4):683-91, 2010 41. Ayhan A, Baykal C, Al A, Ayhan A. No relationship between FHIT expression and clinicopathologic prognostic parameters in early stage cervical carcinoma. Int J Gynecol Cancer, 13(2):192-196, 2003 42. Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature 411:324, 2004 43. Ayhan A, Yasui W, Yokozaki H, ve ark. Loss of Heterozygosity at the bcl-2 Gene Locus and Expression of bcl-2 in human gastric and colorectal carcinomas. Jpn J Cancer Res 85(6):584-591, 1994 44. Deng Y. Telomere dysfunction and tumor suppression: the senescence connection. Nature Rev Cancer 8:450, 2008 45. Yang SX. Bevacizumab and breast cancer: current therapeutic progress and future perspectives. Expert Rev Anticancer Ther 9(12):1715-25, 2009 46. F›dler IJ. The pathogenesis of the cancer metastasis. Nat Rev Cancer 3:1083, 2006 47. Sahai E: Illuminating the metastatic cascade. Nat Rev Cancer 7:737, 2007 48. http://www. metastasis.icr.ac.uk 49. Gray J. Genomics of metastasis. Nature 464:989-990, 2010 50. Khalique L, Ayhan A, Whittaker JC, Singh N, Jacobs IJ, Gayther SA, Ramus SJ. The clonal evolution of metastases from primary serous epithelial ovarian cancers. Int J Cancer 124(7):1579-86, 2009 51. Mc Cawley LJ, Matrisiani LM. Matrix Metalloproteinases: multifunctional contibutors to tumor progression. Mol Med Today, 6:149-156, 2000 52. Ayhan A, Tok EC, Bildirici I, Ayhan A. Overexpression of CD44 variant 6 in human endometrial cancer and its prognostic significance. Gynecol Oncol 80(3):355-8, 2001 53. Ayhan A, Baykal C, Al Atakan, Ayhan A. Prognostic significance of CD44v6 in uterine cervix carcinomas. Gynecol Oncol 83 (3):56974, 2001 54. Ekici S, Ayhan A, Kendi S, Özen H. Determination of prognosis in patients with prostate cancer treated with radical prostatectomy: prognostic value of CD44v6 score. J Urol 167(5):2037-41, 2002 55. Nguyen D, Massague J. Genetic determinants of cancer metastasis. Nat Rev Genet 8:341, 2007 56. Ayhan A, Yasui W, Yokozaki H, Kitadai Y, Tahara E. Reduced expression of nm23 protein is associated with advanced tumor stage and distant metastasis in human colorectal carcinomas. Virchows Arch B Cell Pathol 63:213-218, 1993 57. Saraç E., Ayhan A., Ertoy D., ve ark. nm23 expression in carcinoma of the uterine cervix. Eur J Gynecol Oncol 19 (3):312-315, 1998 58. Ayhan A, Bardak Y, Çekiç O, ve ark.: Nm23 expression in choroidal melanoma. Ophtalmic Res 32(6):257-260, 2000 26 J‹NEKOLOJ‹K ONKOLOJ‹ 59. Ünal VS, Ayhan A, Tokgözo¤lu MA. Proliferating-cell nuclear antigen index and nm23 expression in osteosarcoma in relation to disease-free survival and tumor grade. Saudi Med J 26(9):1475-7, 2005 60. Tumors of the breast and Female Genital Organs. Pathology and Genetics. WHO. Tavassoli FA, Devilee P. (eds). (2003) Lyon: IARC Press 61. Lynch HT, de ka Chapelle A, Hereditary colorectal cancer. NEJM 348:919, 2003 62. Cleaver JE, Lam ET, Revet I. Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat Rev Genet 10(11):756-68, 2009 63. D’Andrea AD. Susceptibility pathways in Fanconi’s anemia and breast cancer. N Engl J Med.362(20):1909-19, 2010 64. Ting A et al. The cancer epigenome-components and functional correlates. Genes Dev 20:3215, 2006 65. Esteller M. Epigenetics in cancer. N Eng J Med 358:1148, 2008 65a Yamanaka S, Blau HM. Nuclear reprogramming to a pluripotent state by three approaches. Nature 465:704, 2010 65b.Pera MF. The dark side of induced pluripotency. Nature 471:46-7, 2011. 66. Kurman RJ, Ellenson LH, Ronnet BM. (Eds) Blaustein`s Pathology of the Female Genital Tract. Springer, 2011 67. Herrington S. Recent advances in molecular gynecologic pathology. Histopathology 55:243-49, 2009 68. Pett M, Coleman N. Integration of high-risk HPV: a key event in cervical carcinogenesis? J Pathol 212:356-67, 2007 69. Kersemaekers AM, Fleuren GJ, Kenter GG, Van den Broek LJ, Uljee SM, Hermans J, Van de Vijver MJ. Oncogene alterations in carcinomas of the uterine cervix: overexpression of the epidermal growth factor receptor is associated with poor prognosis. Clin Cancer Res. 5:577-86, 1999 70. Kim JY, Lim SJ, Park K, Lee CM, Kim J. Cyclooxygenase-2 and cerbB-2 expression in uterine cervical neoplasm assessed using tissue microarrays. Gynecol Oncol 97(2):337-41, 2005 71. Goncalves A, Fabbro M, Lhommé C, Gladieff L, Extra JM, Floquet A, Chaigneau L, Carrasco AT, Viens P. A phase II trial to evaluate gefitinib as second- or third-line treatment in patients with recurring locoregionally advanced or metastatic cervical cancer. Gynecol Oncol 108(1):42-6, 2008 72. Gaffney DK. Winter K, Dicker AP, et al. Efficacy and patterns of failure for locally advanced cancer of the cervix treated with celebrex (celecoxib) and chemoradiotherapy in RTOG 0128. Int J Radiat Oncol Biol Phys 69(1):111-7, 2007 73. Aoyama C, Peters J, Senadheera S, Liu P, Shimada H. Uterine cervical dysplasia and cancer: identification of c-myc status by quantitative polymerase chain reaction. Diagn Mol Pathol 7(6):324-30 1998 74. Segawa T, Sasagawa T, Yamazaki H, Sakaike J, Ishikawa H, Inoue M. Fragile histidine triad transcription abnormalities and human papillomavirus E6-E7 mRNA expression in the development of cervical carcinoma. Cancer. 1;85(9):2001-10, 1999 75. Sotiropoulou N, Bravou V, Kounelis S, Damaskou V, Papaspirou E, Papadaki H.Tumour expression of lymphangiogenic growth factors but not lymphatic vessel density is implicated in human cervical cancer progression. Pathology 42(7):629-36, 2010 76. Monk BJ, Sill MW, Burger RA, Gray HJ, Buekers TE, Roman LD.Phase II trial of bevacizumab in the treatment of persistent or recurrent squamous cell carcinoma of the cervix: a gynecologic oncology group study. J Clin Oncol. 27(7):1069-74, 2009 77. Klaes R, Benner A, Friedrich T et al. P16INK4A immunohistochemistry improves intraobserver agreement in the diagnosis of CIN. Am J Surg Pathol 26: 1389-99, 2004 78. Pinto AP, Crum CP, Hirsch MS. Molecular markers of early cervical neoplasia. Diagn Histopathol (Oxf). 16(10):445-454, 2010 79. Tatum JL, Kelloff GJ, Gillies RJ, et al. Hypoxia: importance in tumor biology, noninvasive measurement by imaging, and value of its measurement in the management of cancer therapy. Int J Radiat Biol. 82(10):699-757, 2006 80. van Seters M, ten Kate FJ, van Beurden M, Verheijen RH, Meijer CJ, Burger MP, Helmerhorst TJ. In the absence of (early) invasive carcinoma, vulvar intraepithelial neoplasia associated with lichen sclerosus is mainly of undifferentiated type: new insights in histology and aetiology. J Clin Pathol. 60(5):504-9, 2007 81. Worsham MJ, Van Dyke DL, Grenman SE, Grenman R, Hopkins MP, Roberts JA, Gasser KM, Schwartz DR, Carey TE. Consistent chromosome abnormalities in squamous cell carcinoma of the vulva. Genes Chromosomes Cancer. 3(6):420-32, 1991 82. Hefler LA, Sliutz G, Leodolter S, Speiser P, Joura E, Reinthaller A, Kohlberger P. Squamous cell carcinoma antigen serum levels as prognostic parameter in patients with early stage vulvar cancer. Gynecol Oncol 97(3):904-7, 2005 83. Knopp S, Tropè C, Nesland JM, Holm R. A review of molecular pathological markers in vulvar carcinoma: lack of application in clinical practice. J Clin Pathol 62(3):212-8, 2009 84. Liu F-S. Molecular carcinogenesis of endometrial cancer. Taiwanese J Obstet Gynecol 46:26-32, 2007 85. www. Endometrium.org.; Hecht JH and Mutter GL. JCO 24:47834791, 2006 86. Llobet D, Pallares J, Yeramian A, et al. Molecular pathology of endometrial carcinoma; practical aspects from the diagnostic and therapeutical viewpoints. J Clin Pathol 62:777-85, 2009 87. Garg K, Soslow RA. Lynch syndrome (HNPCC) and endometrial carcinoma. J Clin Pathol 62:679-684, 2009 88. Fadare O, Zheng W. Insights into Endometrial Serous Carcinogenesis and Progression. Int J Clin Exp Pathol 2:411-432, 2009 89. Zheng W, Xiang L, Fadare O, Kong B. A proposed model for endometrial serous carcinogenesis (Review). Am J Surg Pathol 35(1):e1-e14, 2011 90. Nucci MR and Oliva E. (Eds) Gynecologic pathology. Elsevier Churchill Livingstone 2009 91. Jarboe EA, Folkins AK, Drapkin R et al. Tubal and ovarian pathways to pelvic epithelial cancer: a pathological perspective. Histopathology 53:127-138, 2008 92. Widschwendter M, Apostolidou S,Jones AA, Fourkala EO, Arora R, Pearce LC, Frasco M, Ayhan A, Zikan M, Cibula D, Iyibozkurt CA, Yavuz A, Hauser-Kronberger C, Dubeau L, Menon U and Jacobs IJ. HOXA methylation in normal endometrium from premenopausal women is associated with the presence of ovarian cancer – a proof of principle study. Int J Cancer 25(9):2214-8, 2009 93. Kurman RJ, Shih IeM. The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am J Surg Pathol. 34(3):433-43, 2010 94. Karst AM, Drapkin R.Ovarian cancer pathogenesis: a model in evolution. J Oncol 2010:932371, 2010 95. Singer G, Kurman RJ and Russel P. Diverse tumorigenic pathways in ovarian serous carcinoma. Am J Pathol 160: 1223-28, 2002 96. Veras E, Mao TL, Ayhan A, Ueda S, Lai H, Hayran M, Shih IM, Kurman RJ. Cystic and Adenofibromatous Clear Cell Carcinomas of the Ovary. Distinctive Tumors That Differ in their Pathogenesis and Behavior: A Clinicopathologic Analysis of 122 Cases. Am J Surg Pathol 33:844-53, 2009 97. Kuo KT, Mao TL, Jones S, Veras E, Ayhan A, Wang TL, Glas R, Slamon D, Velculescu VE, Kurman RJ, Shih IM. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am J Pathol 174(5):1597-601, 2009 BÖLÜM 19 ❏ Kanserin Moleküler Temeli ve Jinekolojik Kanserlerde Önemi 98. Sehdev AS, Kurman RJ, Kuhn E, Shih IeM. Serous tubal intraepithelial carcinoma upregulates markers associated with high-grade serous carcinomas including Rsf-1 (HBXAP), cyclin E and fatty acid synthase. Mod Pathol 23(6):844-55, 2010 99. Jones S, Wang TL, Shih IeM, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 330(6001):228-31, 2010 100.Vang R, Shih IM, Salani R, Sugar E, Ayhan A, Kurman RJ. Subdividing Ovarian and Peritoneal Serous Carcinoma into Moderately Differentiated and Poorly Differentiated Does not Have Biologic Validity Based on Molecular Genetic and In Vitro Drug Resistance Data. Am J Surg Pathol 32(11):1667-74, 2008 101.Ayhan A, KurmanRJ, Vang R, Logani S, Seideman J, Shih IM. Defining the cut-point between low- and high- grade ovarian serous carcinomas: A clinicopathologic and molecular genetic analysis. Am J Surg Pathol 33(8):1220-4, 2009 102.Singh N, Ayhan A, Menon U, Chin Aleong JA, Faruqi AZ, Gayther SA, Jacobs IJ. Grading of serous ovarian carcinoma: further evi- 27 dence of a lack of agreement between conventional grading systems. Histopathology 52(3):393-5, 2008 103.Crum, CP and Xian W. Bringing the p53 Signature Into Focus. Editorial. Cancer 116:5519-21, 2010 104.Norquist BM, Garcia RL, Allison KH et al. The molecular pathogenesis of ovarian carcinoma: alterations in tubal epithelium of women with BRCA1 and BRCA2 mutations. Cancer 116:5261-71, 2010 105.Brinkmann D, Ryan A, Ayhan A, McCluggage WG, Feakins R, Santibanez-Koref MF, Mein CA, Gayther SA, Jacobs IJ. A molecular genetic and statistical approach for the diagnosis of dual-site cancers. J Natl Cancer Inst 96(19):1441-1446, 2004 106.Ramus SJ, Elmasry K, Luo Z, Gammerman A, Lu K, Ayhan A, Singh N, McCluggage WG, Jacobs IJ, Whittaker JC, Gayther SA. Predicting clinical outcome in patients diagnosed with synchronous ovarian and endometrial cancer. Clin Cancer Res. 14(18):58408, 2008
© Copyright 2026 Paperzz