Paper
zz
Explore Categories
Log in
Create new account
No category
Selim deri tümörleri ve sistemik malinite ilişkisi
Download
Report
Bu PDF dosyasını indir
2.Bölüm Toprağın Oluşumu
2015 yılı onaylananlar
dosyayı indir
1277 - Ege Üniversitesi Diş Hekimliği Fakültesi
Atipik Goldenhar sendromu
Multipl Miyeloma Eşlik Eden Nekrobiyotik
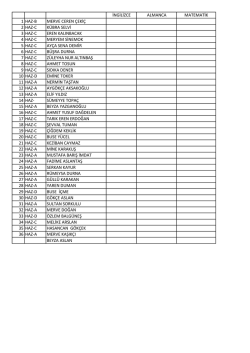
ingilizce almanca matematik 1 haz-b merve ceren çekiç 2 haz
Uvea Tümörlerinde Cerrahi Tedavi
Tıbbi Alan, İncelemesi / Deneyi Yapılan Ürünler veya Malzemeler
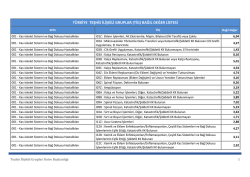
(tig) bağıl değer listesi
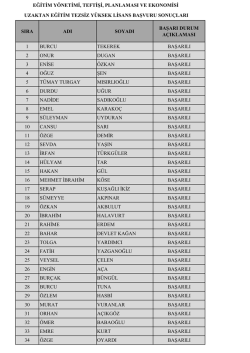
Uzaktan Eğitim Başvuru Sonuçları için tıklayınız
© Copyright 2026 Paperzz
About Paperzz
DMCA / GDPR
Report