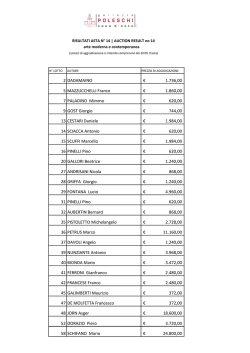
Alberi genealogici e loro complicazioni (Prima parte dell’individuazione dei loci responsabili di patologie genetiche in uomo) Prof. Mario Ventura ATTENZIONE ricordate e tenete presente che lo studio della genetica umana si articola su due linee: Ricerca: individuare i geni che controllano le nostre funzioni, capirne l’ereditarieta’, studiare la nostra evoluzione . Per questi aspetti l’uomo e’ un organismo come gli altri con qualche difficolta’ in piu’ per l’impossibilita’ di fare incroci predeterminati e mutagenesi. Inoltre l’uomo di solito ha poca progenie. Prof. Mario Ventura ATTENZIONE Diagnostica: i risultati della ricerca possono portare alla diagnostica e alla prevenzione delle patologie ereditarie. Ne consegue che se devo studiare un carattere nuovo il modo di procedere e’ diverso da quando devo fare consulenza genetica: se sto ricercando devo procedere verso l’ignoto e scegliero’ le strategie di volta in volta diverse perche’ collegate all’informazioni che ho al momento di partire. se sto facendo consulenza parto da informazioni sul carattere gia’ note e devo concentrami su una particolare famiglia e adottare gli strumenti piu’ utili per quella famiglia Prof. Mario Ventura ATTENZIONE Cominceremo dall’aspetto di ricerca e prenderemo ad esempio di studio alcuni fenotipi patologici per capire come si ragiona quando si vuole studiare una funzione. Quindi cominciamo a richiamare alcune nozioni di base sulle modalita’ di trasmissione e come si studiano gli alberi genealogici che sono la versione umana degli incroci studiati a Genetica di base. Prof. Mario Ventura Autosomica dominante I Una persona che manifesta il carattere ha almeno un genitore con lo stesso fenotipo II Aa aa aa Aa aa aa Aa aa Aa Aa aa Aa Aa III aa Il figlio di chi manifesta il carattere ha il 50% di probabilita’ di ereditare l’allele e di manifestare il fenotipo Entrambi i sessi presentano il carattere Assumendo che il genitore sia eterozigote il che di solito e’ vero per fenotipi rari E’ trasmessa da entrambi i sessi Prof. Mario Ventura Autosomica recessiva Entrambi i sessi presentano il fenotipo Aumentata incidenza della consanguineita’ aa Aa A- ? ? ? A- A- A- Aa Aa Aa Aa Aa aa aa aa ? ? A- A- ? aa Gli individui che presentano il fenotipo di solito sono figli di soggetti che non presentano il fenotipo aa A- aa Ciascun figlio successivo a quello che presenta il carattere ha il 25% di probabilita’ di mostrarlo (I legge di Mendel) INDIVIDUI CON UN FRATELLO O SORELLA CON IL CARATTERE HANNO 2/3 DI PROBABILITA DI ESSERE PORTATORI DELL ALLELE MUTATO. Prof. Mario Ventura X-linked recessiva ? Esprimono il carattere quasi esclusivamente i maschi I genitori di solito non presentano il carattere soprattutto se e’ una patologia grave. XaY X AX a Lo stato di portatrice della madre e’ certo se ha il padre o fratelli che manifestano (nel caso delle patologie gravi ? il padre non e’ mai affetto) ? ? ? ? XaY Le femmine possono manifestare il carattere: 1. se il padre lo manifesta il carattere e la madre eterozigote Prof. Mario Ventura 2. se l’inattivazione del cromosoma X non e’ casuale X-linked dominante Entrambi i sessi presentano il carattere. Se e’ una patologia le femmine presentano un fenotipo meno grave dei maschi per effetto dell’inattivazione I figli di una donna che manifesta hanno il 50% di essere affetti, indipendentemente dal sesso Un maschio che manifesta avra’ tutte le figlie che manifestano e tutti i figli maschi senza il carattere Prof. Mario Ventura Y -linked solo i maschi I maschi che manifestano hanno il padre con lo stesso fenotipo Tutti i figli maschi di un uomo che manifesta, manifesteranno il fenotipo NON SI CONOSCONO GENI-MALATTIA LEGATI AL CROMOSOMA Y Prof. Mario Ventura Elevata frequenza dell’allele recessivo e’ un carattere dominante? OO Prof. Mario Ventura AO AA AO AA AO OO AO AO OO OO AO AO AO OO OO Concepiti maschi abortiti Dominante X linked Prof. Mario Ventura Eredita’ mitocondriale Figure 3.4. Pedigrees of mitochondrial diseases. (A) A typical pedigree pattern, showing mitochondriallydetermined hearing loss (family reported by Prezant et al., 1993). Sordita’ B) The atypical pattern of Leber's hereditary optic atrophy, which affects mainly males (family reported by Sweeney et al., 1992). Prof. Mario Ventura Atrofia ottica ereditaria di Leber (LHON) Eredita’ mitocondriale Eredita’ matrilineare: - interessa entrambi i sessi; - trasmesse da madri affette Eteroplasmia ed omoplasmia Prof. Mario Ventura Nuova mutazione Qual e’ il modello piu’ probabile di ereditarieta’? mutazione DOMINANTE Prof. Mario Ventura Ancora complicazioni SPESSO LA MALATTIA SI PRESENTA IN FORMA SPORADICA aa aa aa NUOVA MUTAZIONE: nessuno dei genitori porta la mutazione; essa si verifica in uno dei gameti che da origine all individuo affetto. Questo sembra essere il caso di forme gravi di OI, che non sono generalmente compatibili con la riproduzione. In questo caso il rischio di avere un altro figlio affetto e praticamente nullo……a meno che non si Aa tratti di MOSAICISMO Prof. Mario Ventura Mosaicismo MOSAICISMO SOMATICO O DELLA LINEA GERMINALE: un individuo presenta alcune cellule mutate e altre no. Se le cellule mutate sono in prevalenza rispetto a quelle wild-type si possono manifestare segni clinici della malattia. La mutazione e insorta durante lo sviluppo embrionale. Piu precocemente insorge maggiore e il numero di cellule mutate. ESISTE UN RISCHIO DI TRASMISSIONE DELLA MALATTIA. Anche se non quantizzabile. Questo sembra spiegare casi di famiglie con piu di un figlio affetto, ma genitori sani e non portatori della mutazione (se nota). Prof. Mario Ventura Mosaicismo germinale CSp=Controllo sperma PSp=Paziente sperma N OI CSp PSp 225 nt 72 nt 63 nt PCR coni geni per COL1A1 la mutazione e’ presente nelle cellule germinali del padre Prof. Mario Ventura Mosaicismo germinale e chimerismo Mosaico Chimera Il chimerismo e’ un fenomeno piuttosto raro, diversamente dal mosaicismo Prof. Mario Ventura Eterogeneita’ genetica Questo concetto si applica alla popolazione ed e’ un indice della nostra ignoranza dei processi che portano all’espressione del fenotipo non esiste corrispondenza 1:1 gene:fenotipo Eterogeneita’ di locus Il fenotipo e’ il risultato di una catena di interazioni: il risultato finale puo’ risultare simile anche quando i loci sono diversi. Eterogeneita’ allelica/Serie allelica Fenotipi diversi possono essere originati da mutazioni in siti diversi dello stesso gene o da tipi diversi di mutazione a carico dello stesso gene. Prof. Mario Ventura Eterogeneita’ genetica di locus (geni diversi = fenotipo) Il fenotipo puo’ essere molto lontano dal gene e dal suo prodotto primario in quanto esso risulta dall’interazione di prodotti genici. Il primo passo e’ definire le modalita’ di trasmissione: Uno stesso fenotipo puo’ mostrare tipi di ereditarieta’ diversa (questo immediatamente segnala la presenza di piu’ loci). Retinite pigmentosa 6 locus X-linked, ~20 locus autosomici trasmessi sia come caratteri recessivi che dominanti Alterazioni del collagene di tipo1: ci sono 2 loci dal momento che questo collagene e’ formato da un trimero: il fenotipo viene comunque identificato come Osteogenesi imperfetta Prof. Mario Ventura Eterogeneita’allelica/Serie allelica (un solo gene diversi fenotipi) Fenotipi diversi possono essere originati da mutazioni in siti diversi dello stesso gene o da tipi diversi di mutazione. AR (gene per il recettore per gli androgeni) : la mutazione puntiforme che provoca l’inattivazione del gene origina la femminilizzazione testicolare. L’espansione di triplette l’atrofia muscolo-spino-bulbare Ret (recettore tirosi-chinasi): Mutazioni che provocano la perdita di funzione originano la sindrome di Hirschsprung (sindrome del megacolon). Mutazioni puntiformi che generano un aumento di funzione originano tumori familiari delle ghiandole endocrine MEN (tidoide e adrenali). Altre mutazioni portano entrambe PMP22: la duplicazione di una regione di 1.5 Mb provoca CMT1A,distrofia muscolare neurodegenerativa con ipomielinizzazione delle fibre nervose. La delezione dello stesso gene porta ad HNPP: sindrome neurologica con paralisi e ipermielinizzazione Prof. Mario Ventura Penetranza Nel caso dei caratteri mendeliani classici il fenotipo corrisponde esattamente al genotipo (compatibilmente con la dominanza e la recessivita’). Spesso pero’ a livello del singolo individuo questo puo’ non essere vero o immediamente evidente. Un carattere presenta penetranza pari al 100% quando tutti i portatori di un certo allele manifestano il fenotipo corrispondente: pisello verde o giallo, colore bianco, rosso, rosa o striato dei fiori, nanismo acondroplasico nell’uomo, gruppo sanguigno. Un carattere presenta penetranza pari al 70% quando solo il 70% degli individui portatori dell’allele manifestano il fenotipo. Prof. Mario Ventura Penetranza La penetranza e’ quindi un concetto che si riferisce alla popolazione. A livello del singolo individuo il carattere ha solo due possibilita’: si manifesta o non si manifesta. E’ piu’ frequente nei caratteri dominanti ed e’ piu’ evidente nella genetica umana perche’ la genetica umana si applica ad una popolazione “selvatica”. Sapere che un gene puo’ non essere completamente penetrante, e’ critico per studiarne la genetica o fornire consulenza genetica: un certo soggetto che non manifesta il carattere puo’ essere portatore del gene. Prof. Mario Ventura Maggiore difficolta’ in consulenza genetica!!! Ancora complicazioni: Insorgenza tardiva penetranza correlata con l’eta’ Non tutti i disordini genetici sono congeniti Molti sono congeniti e genetici ... ma non tutti il fenotipo puo’ comparire in eta’ avanzata dal punto di vista genetico raramente questi fenotipi sono dovuti a nuove mutazioni a livello di popolazione l’allele mutato puo’ essere frequente purche’ l’insorgenza si verifichi dopo l’inizio dell’eta’ riproduttiva e non limiti la fitness FITNESS: IDONEITA’ BIOLOGICA = capacita’ di avere figli Prof. Mario Ventura Insorgenza tardiva Curve relative all’eta’ di insorgenza della Corea di Huntigton. A. probabilita’ che un individuo che porta il gene responsabile della malattia ne abbia presentato i sintomi ad una certa eta’. B. rischio che il figlio sano di un genitore affetto possegga il gene della malattia ad una data eta’. MOLTE patologie mitocondriali sono ad insorgenza tardiva Prof. Mario Ventura Anticipazione E’ la tendenza di alcun condizioni dominanti ad espressivita’ variabile a diventare piu gravi con le generazioni successive MOTIVO? espansione di triplette: tipo di mutazione individuata nel 1990 Corea di Hunghinton, X fragile, Distrofia Miotonica premutazione: stato che aumenta il rischio di trasmettere il fenotipo patologico. Il rischio non e’ piu’ quantizzabile in termini probabilistici Prof. Mario Ventura Espansioni di triplette instabili:mutazioni dinamiche Mutazioni dovute all’espansione di trinucleotidi localizzate nelle regioni codificanti o non-codificanti di alcuni geni, si originano per errori nella replicazione del DNA. Possono originare un sito fragile visibile citogeneticamente (sindrome del sito fragile del chrX = FRAX). Prof. Mario Ventura Meccanismo di formazione dell’espansione Prof. Mario Ventura Espansioni di triplette instabili:mutazioni dinamiche Se si trova in una regione codificante il prodotto del gene presenterà n numero di residui amminoacidici corrispondenti alla tripletta superiori al prodotto dell’allele selvatico. Di solito la tripletta coinvolta è CAG (acido glutammico). Se si trova in una regione non-codificante possono alterare l’espressione del gene in cis. Distrofia miotonica: 3’UTR; atassia di Friedrich: introne1; X-fragile:5’UTR. In genere il numero di triplette associato allo stato di mutazione piena è più alto se l’amplificazione colpisce regioni non tradotte, ma comunque importanti per la trascrizione o per il trascritto Prof. Mario Ventura Sindrome della X-fragile Prof. Mario Ventura Schema di mutazioni dinamiche Mutazione completa, range di mutazione CGG1000 GAA900 CTG3000 CGG GAA CTG CGG CGG 280 CGG CG 200 G 60 Premutazione GAA 200 GAA GAA CGG GAA GAA GAA 6 GAA 5’UTR CTG CTG CTG GAA 22 GAA CGG CGG CGG 52 CGG Normale CTG GAA esone X-fragile CTG 50 CTG CTG CTG 37 CTG 7 introne CTG esone introne Atassia di Friedrich Mutazione completa, range di mutazione CAG121 Premutazione CAG 35 CGG 25 CAG CGG CAG CGG CAG 36 CAG CAG esone 3’UTR Distrofia miotonica CGG75 CAG CAG Normale 3 CGG 10 CGG CGG Esone ORF 49 7 Atassia spinocerebellare 1 Corea di Huntington Prof. Mario Ventura Analisi delle mutazioni da amplificazione di triplette Primer Normale 5-50 Premutazione 50-200 Mutazione completa >200 Prof. Mario Ventura
© Copyright 2026 Paperzz