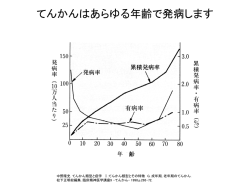
小児の精神運動発達 脳神経系の発達・神経学的診察法 小児の神経・筋疾患 M3 講義 脳性麻痺 受胎から生後4週までに生じた脳の非進行性病変に基 づく、永続的な しかし変化しうる運動・姿勢の異常と 定義され、進行性疾患や将来正常化するであろうと 思われる運動発達遅延は除外する (厚生省1968年) 脳性麻痺は運動・姿勢の異常であり、知能障害の有無 は関係無い。 脳性麻痺の分類 脳性麻痺の原因 運動障害による分類 1 痙直型 2 アテトーゼ型 3 強剛型 4 失調型 5 低緊張型 6 混合型 出生前 脳形成障害、胎内感染、頭蓋内出血、先天性水 頭症など 障害の分布による分類 周生期・新生児期 新生児仮死など低酸素性虚血性脳症、脳室周 囲白質軟化症(PVL)、頭蓋内出血、高ビリルビン血 症(核黄疸)、中枢神経感染 単麻痺 片麻痺 四肢麻痺 全般的な知的機能の障害 社会生活への適応障害 18歳未満に発症する (境界域: およそ70 ~ IQ 79) 軽度精神遅滞 : IQ 50-55 ~ およそ70 中等度精神遅滞 : IQ 35-40 ~ 50-55 重度精神遅滞 : IQ 20-25 ~ 35-40 最重度精神遅滞 : IQ 20-25 以下 両片麻痺 両麻痺 三肢麻痺 代表的な発達・知能検査 精神遅滞(知的障害) 対麻痺 遠城寺式乳幼児分析的発達評価表(4歳半まで) 日本語版デンバー式発達スクリーニング検査 (6歳まで) 津守・稲毛式発達検査(7歳11か月まで) ウエクスラー式知能検査 WIPPSI (3歳10か月~7歳1か月) WISC-III (5歳~16歳11か月) K式発達検査(13歳4か月まで) 田中・ビネー知能検査(2歳~成人) 1 精神遅滞をきたす代表的疾患(1) 精神遅滞をきたす代表的疾患(2) 出生前の原因 染色体異常 Down症候群(trisomy 21)、脆弱X症候群など 奇形症候群・脳奇形 Prader-Willi症候群、Sotos症候群、滑脳症など 代謝異常症 ムコ多糖症、Lesch-Nyhan症候群、Menkes病など 外 因 有機水銀、薬物、アルコール、先天感染(風疹やサ イトメガロウィルスなど)etc 周生期の原因 新生児仮死、MASなどの低酸素性虚血性脳症 頭蓋内出血、中枢神経感染、低血糖、高ビリルビン血症 てんかん (epilepsy) てんかんの定義 大脳ニューロンの過剰な突発的発射に由来する反 復性(2回以上)の発作を主徴とする慢性の脳疾患。 ※大脳ニューロンの過剰な発射に由来しない い わゆる状況関連性発作を除外する。 てんかんと見誤りやすいもの (小児) 熱性けいれん、息どめ発作 チック 軽症下痢に伴う発作 失神 睡眠時(入眠時)ぴくつき、悪夢 心因発作 急性代謝障害(低血糖、テタニー) かんしゃく 小児のてんかん West症候群(点頭てんかん) 乳児期に好発。シリーズを形成するトニック・スパズム。 脳波:ヒプスアリスミア 治療:抗てんかん薬(バルプロ酸、Vit.B6など)、ACTH療法。 神経学的予後:不良。レノックス(Lennox)症候群への移行 出生後の原因 頭部外傷、中枢神経感染・脳症、低酸素性虚血性脳症 難治性てんかん(West症候群、Lennox-Gastautなど) 脳血管障害、慢性硬膜下血腫 心因性(愛情遮断などの虐待、劣悪な環境)、低栄養など その他:自閉症、難聴 原因不明の知的障害 てんかん発作の分類診断 (→カルバマゼピン) Ⅰ. 部分発作 A 単純部分発作 B 複雑部分発作 C 部分発作から全般性強直・間代発作へ移行する発作 (→バルプロ酸) Ⅱ. 全般発作 A 欠神 B ミオクロニー C 間代 D 強直 E 強直・間代 F 脱力 Ⅲ. 分類不能 小児欠神てんかん 好発年齢:6-7歳がピーク。女児に多い。 5-20秒前後の意識消失発作を頻発。 脳波:3Hzの全般性棘徐波複合。バルプロ酸が有効。 思春期頃に全般性強直発作が出現することあり。 2 筋トーヌスを評価する その他の小児てんかん 良性新生児てんかん 若年性欠神てんかん 若年性ミオクローヌスてんかん 覚醒時大発作てんかん 中心・側頭部に棘波を有する良性小児部分てんかん Lennox-Gastaut症候群 早期乳児てんかん性脳症 筋トーヌス(筋緊張) 骨格筋は不随意にたえず緊張した状態にあり、この緊張 を筋トーヌスをよぶ。 休止時の筋緊張(受動的筋緊張) 1. Passivity(被動性:筋の振れ具合) 2. Extensibility(伸展性:筋の伸び具合) 3. Consistency(筋の柔らかさ) 姿勢保持の筋緊張(能動的筋緊張) 運動時筋緊張 筋緊張亢進:錐体路(痙性)、錐体外路(固縮)など 筋緊張低下:錐体路、小脳、後索、末梢神経、筋 floppy infantの鑑別診断 floppy infant 生下時より筋緊張が低下し、グニャグニャした感じのする小児。 筋力低下(+) 抗重力運動ができない DTR(-)~(±) 蛙肢位姿勢 筋力低下(ー) 抗重力運動ができる DTR(+) 神経原性 脊髄性筋萎縮症 (Werdnig-Hoffmann病, SMA I) 筋原性 先天性筋ジストロフィー 福山型、非福山型など 先天性筋強直性ジストロフィー 先天性ミオパチー ネマリンミオパチー、中心核病など 代謝病(糖原病、ミトコンドリア病) 中枢神経系異常 非進行性:知的障害、脳性麻痺、脳外傷 進行性:変性疾患 その他 Head lag 陽性 逆U姿勢 二つ折り現象 小児に多い筋疾患の分類 染色体異常など: Down症候群、Prader-Willi症候群 結合織疾患:Ehlers-Danlos症候群 甲状腺機能低下、先天性良性筋緊張低下 筋ジストロフィーの筋病理像 壊死・再生像 1)筋ジストロフィー Duchenne型筋ジストロフィー Becker型筋ジストロフィー 肢帯型筋ジストロフィー 顔面肩甲上腕型筋ジストロフィー 先天性筋ジストロフィー 福山型先天性筋ジストロフィー 非福山型先天性筋ジストロフィー 2)先天性筋強直性ジストロフィー 3)先天性ミオパチー ネマリンミオパチー セントラルコア病 中心核病 4)ミトコンドリア病 5)筋炎 多発性筋炎 皮膚筋炎 オパーク細胞 筋の大小不同 線維化 脂肪置換など 3 DMDの臨床症状 ジストロフィンの異常 Duchenne型筋ジストロフィー(DMD) Becker型筋ジストロフィー(BMD) (BMDはDMDよりも軽症で、15歳で歩行可能。) X連鎖劣性遺伝(Xp21) 原因遺伝子:ジストロフィン DMDは筋ジストロフィーの中で最も頻度が高い。 男児 2,000~3,000人に1人 (BMDはその半分程度) 進行性の運動障害 新生児・乳児期に特に異常はない 1歳6か月:独歩の遅れが30-50% 3~5歳 :転びやすい、走れない、階段を昇れない 動揺性歩行(waddling gait) 登はん性起立(Gowers徴候) 下腿の(仮性)肥大 CK(AST, ALT, LDH)上昇 (数倍~数十倍) (足)関節の拘縮、尖足 10歳前後:歩行不能(車いす) 15歳以降:呼吸不全(非侵襲的陽圧換気の導入) :心不全 平均寿命 :28-30歳前後 (以前は20歳前後) IQは平均80前後で、軽度~中等度の知的障害は稀でない。 福山型先天性筋ジストロフィー 責任遺伝子: fukutin (9p31) (3’非翻訳領域の3kbレトロトランスポゾンの挿入) 症 状:出生時から全身の筋緊張低下、 筋萎縮、顔面筋罹患 特 徴:日本人に多い、多発関節拘縮、精神遅滞 脳形成障害(大脳・小脳の多小脳回) 登はん性起立(Gowers徴候) 下腿の(仮性)肥大 先天性筋強直性ジストロフィー 先天性ミオパチー 新生児・乳幼児期早期からの筋力、筋緊張低下があり、病理学的に特徴ある所見を 呈する疾患群。多くは歩行を獲得するが、以後も筋力低下は持続する。 ・重症型 (severe infantile form) ・良性先天型 (benign/moderate congenital) ・成人発症型 (adult onset form) ネマリン ミオパチー 遺 伝 常染色体優性 常染色体劣性 X連鎖劣性 + + セントラル コア病 + + 中心核病 + + + CFTD + + その他の ミオパチー + 筋肉特異構造 を示さないもの + + 発育・発達の遅れ + + + + + + 筋力低下 外眼筋 顔面筋 咽頭・舌・頸筋 近位筋優位 + + + + + + + + + + + + + + + + + + + + + + + + + + + + + + + + + + - +*4 + - + + 筋 病 理 筋線維の大小不同 タイプ1線維優位 小径タイプ1線維 重症例の存在 責任遺伝子:DMPK遺伝子(19p13.2)の3’末端CTGリピート 常染色体優性遺伝(AD) 表現促進現象あり 症状:新生児期から筋力と筋緊張低下、呼吸障害 (しばしば人工呼吸管理が必要とされる。) 口を常に半分あげて表情に乏しい(顔面筋罹患) 全例で精神遅滞を伴う。 筋緊張低下は成長とともに徐々に回復。 ほとんどは筋強直性ジストロフィーの母親から生まれるため、まずは母 親の筋強直を確認。次に遺伝子検査となる。 + 4 脊髄性筋萎縮症 遺伝形式:5番染色体q12-q13 survival motor neuron (SMN)遺伝子 neuronal apoptosis inhibitory protein (NAIP)遺伝子 脊髄前角細胞障害で発症 症状:出生時から成人までにみられる筋力低下、筋緊張低下、舌の線維性筋攣縮 生後6か月時までに発症した場合、 Werdnig-Hoffmann病(SMA type I)という。 検査所見: 血清クレアチンキナーゼ(CK)値が正常上限の10倍以下 筋生検で小角化線維 群萎縮(グループアトロフィー) 筋電図で神経原性変化 結節性硬化症 病初期に比較的みられにくい所見 中枢神経機能障害 関節拘縮症 外眼筋、横隔膜、心筋の障害 聴覚障害 著しい顔面筋罹患 知覚障害 診断基準 常染色体優性遺伝(AD) 約6,000人に1人 (60%以上は孤発例) 全身の多発性過誤腫症 てんかん、知的障害、血管線維腫が3主徴 そろわない人も多い→診断基準を参照。 責任遺伝子 TSC1: hamartin (9q34) TSC2: tuberin (16p13.3) Keywords: 葉状白斑、てんかん(点頭てんかん)、心臓腫瘍、頭蓋内石灰化(上衣下結 節、皮質結節)、知的障害など (http://www.nanbyou.or.jp/sikkan/024_i.htm) Major Features Facial angiofibroma or forehead plaque/顔面の血管線維腫,または前額部 plaque Nontraumatic ungual or periungual fibroma /外傷性ではない爪下,あるいは爪周囲線維腫 hypomelanotic macules (more than 3) /3個以上の色素脱出斑 Shagreen patch / connective tissue nevus /シャグリン斑(粒起革様皮)(結合織母斑) multiple retinal nodular hamartomas /多発性の網膜結節(過誤腫) Cortical tuber /皮質結節 Subependymal nodule /脳室上衣下結節 Subependimal giant cell astrocytoma /脳室上衣下巨細胞星細胞腫 Cardiac rhabdomyoma,single or multiple/1個以上の心臓の横紋筋腫 Lymphangiomyomatosis /リンパ管血管筋腫症 Renal angiomyoma /腎血管筋脂肪腫 白斑(葉状白斑) 顔面の血管線維腫 Minor Features Multiple, randomly distributed dental enamel pits /多発性,散在性の歯エナメル質における小孔 Hamartomatous rectal polyps /過誤腫性の直腸ポリープ Bone cyst /骨嚢胞 Cerebral white matter radial migration lines/大脳白質の放射線状遊走線 Gingival fibromas /歯肉の線維腫 Nonrenal hamartoma /腎以外の過誤腫 Retinal achromic patch /網膜色素脱出斑 Confetti skin lesions /Confetti様皮膚 Multiple renal cysts /多発性腎嚢胞 爪の線維腫 Definitive TSC : either 2 major features or 1 major feature plus 2 minor features Probable TSC : 1 major plus 1 minor feature. Possible TSC : either 1 major features or 2or more minor features. (http://www.geocities.co.jp/SweetHome-Ivory/4475/ts.htm) 4歳 女児 MRI Flair画像 皮質結節 治 療 病変の重症度により異なる。 10か月女児 CT 上衣下結節 14歳 女児 造影CT 腎血管筋脂肪腫 腎嚢胞 心臓腫瘍:主に経過観察 てんかん:点頭てんかん→ACTH療法など その他のてんかん→抗てんかん薬投与 皮膚病変:外科的切除、レーザーなど 腎病変:経過観察、外科的摘出や塞栓術なども。 5 Sturge-Weber症候群 腫瘍病変の発症年齢 顔面の血管腫と同側大脳の脳軟膜血管腫 病因は不明 胎生期の血管叢の異常→脳軟膜の異常血管腫→同部位の静脈血うっ滞→脳皮 質の虚血性障害(萎縮、石灰化)→麻痺、てんかん 臨床症状 顔面の血管腫:三叉神経第1枝領域が中心 脳軟膜血管腫:同側がほとんど けいれん:反対側の部分発作 精神遅滞 眼症状:緑内障、脈絡膜血管腫など Keywords:三叉神経領域の(ポートワイン様)血管腫、同側の脳軟膜血管腫、対 側の部分てんかん 神経線維腫症Ⅰ型 片側の三叉神経領域に血管腫(単純性血管 腫、赤ブドウ酒状血管腫)* (Recklinghausen病, NF-1) 頭部CT 左前頭部、頭頂から後頭部にかけて皮質 を中心に石灰化がみられる。 左大脳半球の萎縮。 * 二重輪郭を示す特徴的な 石灰化像 (rail-road track) カフェ・オ・レ斑、神経線維腫を主徴 とし、骨病変、眼病変、神経腫瘍、 そのほか多彩な症候を呈する全身 性母斑症 。 常染色体優性遺伝(AD) 責任遺伝子:neurofibromin (17q11.2) * 認定基準 NF-1 出 典 1 主な症候 (1) カフェ・オ・レ斑 扁平で盛り上がりのない斑であり,色は淡いミルクコーヒー色から濃い褐色に至るまで様々で,色素斑内に色の濃淡は みられない。形は長円形のものが多く,丸みを帯びたなめらかな輪郭を呈している。 (2) 神経線維腫 皮膚の神経線維腫は思春期頃より全身に多発する。このほか末梢神経内の神経線維腫(nodular plexiform neurofibroma),びまん性の神経線維腫(difuse plexiform neurofibroma) がみられることもある。 2 その他の症候 ① 骨病変-脊柱・胸郭の変形,四肢骨の変形,頭蓋骨・顔面骨の骨欠損など。 ② ③ ④ ⑤ ⑥ ⑦ 眼病変-虹彩小結節(Lisch nodule),視神経膠腫など。 皮膚病変-雀卵斑様色素斑,有毛性褐青色斑,貧血母斑,若年性黄色内皮腫など。 脳脊髄腫瘍-脳神経ならびに脊髄神経の神経線維腫,髄膜腫,神経膠腫など。 脳波の異常 クロム親和性細胞腫 悪性神経鞘腫 3 診断上のポイント カフェ・オ・レ斑と神経線維腫がみられれば診断は確実である。小児例(pretumorous stage)では,径1.5cm以 上のカフェ・オ・レ斑が6個以上あれば本症が疑われ,家族歴その他の症候を参考にして診断する。ただし 両親ともに正常のことも多い。成人例ではカフェ・オ・レ斑が分かりにくいことも多いので,神経線維腫を主 体に診断する。 以上、難病情報センター(http://www.nanbyou.or.jp/sikkan/050.htm)より抜粋。 臨床のための筋病理(埜中征哉著・日本医事新報社) Super Hospital 小児科(梶田光春編集・中山書店) 先天奇形症候群アトラス(梶井正・他、南江堂) 小児疾患の診断治療基準(東京医学社、2001) 必修小児科学アトラス(南江堂) 必修小児科学(南江堂) 臨床小児神経学(前川喜平著、南山堂) 日本てんかん学会HP http://square.umin.ac.jp/jes/ 筋疾患のいろいろ http://www.ncnp.go.jp/hospital/news/kintop.html 小児では、カフェ・オ・レ斑を「思春期前は0.5cm以上、思春期後は1.5cm以上が6個」とすることが多い。 6
© Copyright 2026 Paperzz