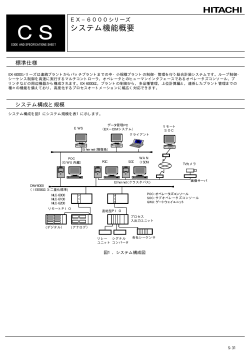
創薬開発において 早期 POC 確保するための手引き 創薬系バイオベンチャーが 早期に臨床試験を開始するための解説書 首都圏バイオネットワーク 事務局: 財団法人バイオインダストリー協会 委託先: POC クリニカルリサーチ株式会社 はじめに 財団法人バイオインダストリー協会の統計調査によると 2006 年末現在の我が国のバイ オベンチャー企業数は 586 社とされており、このうち医薬品、医療機器、診断薬等の開 発企業は 157 社とされており、企業数は毎年増加傾向にある 1)※。 (※詳細は P.47 参考 文献等を参照) しかし、この企業数増加の活況に反し、各社の事業は予断を許さない状況が続いている。 2006 年末までに、14 社のバイオベンチャーの株式上場が達成されたが、近年、その 勢いは衰え、2006 年はファーマフーズ 1 社のみの株式上場となり、2007 年はジーエ ヌアイ社及びジャパン・ティッシュ・エンジニアリング社の 2 社の株式上場に留まって いる。また、2005 年から低下を続けていた上場バイオベンチャーの株価時価総額は、 一時は持ち直したかに見えたが、その後落ち込みから回復できない状態が続いており、 2007 年後半の米国サブプライム問題の我が国市場への影響も災いし、今後も楽観的な 予測には慎重にならざるを得ない状況である。 しかし、このような厳しい環境においても、2008 年 3 月にはナノキャリア社及びカル ナバイオサイエンス社の 2 社が株式上場し、2007 年 12 月から 2008 年 3 月までの 4 箇月間に 3 社の株式上場が達成されている。国内バイオベンチャーの再活況化の到来も そう遠くはないと思われることから、今後のバイオベンチャー業界の動向には注目してい きたいところである。 また、既に臨床開発のステージに入り、ライセンス活動を活発化し、株式上場準備の最終 段階に入ってきている創薬系バイオベンチャーも少なくないことから、これらの上場準備 段階にある企業の更なる発展も期待したいところである。 そして、株式上場準備段階にある創薬系バイオベンチャーにおいては、創薬開発における “Proof of Concept(以下、POC)”を確保しているか、しつつあることからも、創薬開 発の POC を確保するための臨床試験をいかに早く開始できるかが、いかに重要であるか ということが良く分かる。 本書では、この POC 臨床試験を早期に始めるためのポイントを示したい。そして、バイ オテクノロジー応用医薬品、医療機器、診断薬等の研究開発をされているベンチャー企業 の方々が今後の研究開発を検討するうえでの一助になれば幸いである。 2008 年 3 月 首都圏バイオネットワーク事業推進委員 小澤 健夫(POC クリニカルリサーチ株式会社) 2 ヒト POC に関する勉強会 財団法人バイオインダストリー協会が事務局を務め、経済産業省関東経済産業局と連携の 上で推進する首都圏バイオネットワークにおいて、我が国の創薬系バイオベンチャー支援 の活動の一環として「ヒト POC に関する勉強会」が企画され、さまざまなバイオベンチ ャーの創薬開発にかかわる問題が議論されてきた。本勉強会は、バイオベンチャーによる 臨床開発の経験や問題解決事例等の情報を広く共有することにより、各ベンチャー企業の 開発に相互に役立てていただき、我が国の創薬バイオビジネス領域の底上げを図ることを 目的としてきた。すでに株式上場を果たしている企業、これからリードプロダクトの非臨 床試験を開始する企業と、その開発ステージはさまざまであったが、当局の審査経験のあ る先生方や製薬企業の開発経験者にも講師として参加いただき、大変貴重な情報交換がで きたのではないかと考える。また、バイオベンチャー企業間におけるネットワーク形成に も貢献できたのではと考えている。 本書を発刊するきっかけは、この「ヒト POC に関する勉強会」での貴重な議論があり、 このような活動や議論内容をさらに多くの方々にも知っていただき、創薬開発に役立てて いただきたいとの思いからである。 以下に、これまでの「ヒト POC に関する勉強会」のテーマ、講師及びこれまでご参加い ただいた企業等を示す。 本勉強会を支えていただいた方々及びご参加いただいたバイオベンチャー企業の方々に、 改めて心より感謝申し上げたい。 第1回 日時: 2007 年 3 月 15 日(木) 18:30~20:40 講演: 「POC 確保の課題と解決方法」 POC クリニカルリサーチ株式会社 代表取締役社長 小澤 健夫 参加企業: 株式会社イミュノフロンティア、オンコセラピー・サイエンス株式会社、 ナノキャリア株式会社、ワイズセラピューティックス株式会社 第2回 日時: 2007 年 7 月 24 日(火) 19:30~21:45 講演: 「新薬開発と規制当局との付き合い方」 東京大学大学院薬学系研究科 薬学部 医薬品評価科学 准教授 小野 俊介 3 参加企業: 株式会社 Argenes、株式会社イミュノフロンティア、株式会社エムズサ イエンス、オンコセラピー・サイエンス株式会社、株式会社 GBS 研究 所、ディナベック株式会社、ナノキャリア株式会社、株式会社レクメド、 ワイズセラピューティックス株式会社 第3回 日時: 2007 年 10 月 22 日(月) 17:00~19:30 講演: 「CRO との付き合い方」 1) 国内編 アステラス製薬株式会社 開発本部 臨床管理部 課長 津田 郁 2) 海外編 照隅ファルマ株式会社 医薬開発部長 脇 豊 参加企業: 株式会社 Argenes、アリジェン製薬株式会社、株式会社イミュノフロン ティア、株式会社エムズサイエンス、オンコセラピー・サイエンス株式 会社、株式会社 GBS 研究所、ディナベック株式会社、ナノキャリア株 式会社、株式会社レクメド、ワイズセラピューティックス株式会社 第4回 日時:2007 年 12 月 14 日(金) 17:00~19:30 講演:「新薬開発と規制当局との付き合い方(第2回) 」 社団法人日本医師会 治験促進センター 研究事業部 部長 小林 史明 参加企業: アリジェン製薬株式会社、株式会社イミュノフロンティア、オンコセラ ピー・サイエンス株式会社、オンコリスバイオファーマ株式会社、株式 会社 GBS 研究所、株式会社レクメド 4 略語一覧 略 定 語 義 省略していない表現 日本語表現 ADME Adsorption, Distribution, Metabolism, Excretion 吸収・分布・代謝・排泄 ANDA Abbreviated New Drug Application 簡略新薬申請 cGMP Current Good Manufacturing Practice 医薬品の製造に関する基準(米国) CMC Chemical, Manufacturing and Control 化学・製造・品質管理 CMO Contract Manufacturing Organization 製造業務受託機関 CRO Contract Research Organization 開発業務受託機関 DNA Deoxyribonucleic acid デオキシリボ核酸 EC Ethics Committee 倫理委員会 EMEA European Medicines Agency 欧州医薬品庁 EU European Union 欧州連合 FDA Food and Drug Administration 米国食品医薬品局 GCP Good Clinical Practice 医薬品の臨床試験の実施の基準 GLP Good laboratory practice 医薬品の安全性試験の実施に関する基準 GMP Good Manufacturing Practice 医薬品の製造に関する基準 HBV Hepatitis B Virus B 型肝炎ウイルス HCV Hepatitis C Virus C 型肝炎ウイルス HIV Human Immunodeficiency Virus ヒト免疫不全ウイルス H5N1 Hemagglutininn 5、Neuraminidase 1 (トリインフルエンザウイルスの型) IBC Institutional Biosafety Committee 施設内生物学的安全性委員会 ICH International Conference on Harmonization 日米 EU 医薬品規制調和国際会議 IND Investigational New Drug 研究新薬、治験薬 IRB Institutional Review Board 施設内治験審査委員会 MD Medical Doctor 医師 M&A Mergers and Acquisitions 合併と買収 NDA New Drug Application 新薬承認申請 NIH National Institute of Health 米国国立衛生研究所 PMDA Pharmaceuticals and Medical Devices Agency 独立行政法人医薬品医療機器総合機構 POC Proof of Concept 開発コンセプトの妥当性を傍証すること 5 目次 はじめに .......................................................................................................................................................................... 2 ヒト POC に関する勉強会.................................................................................................................................... 3 略語一覧 .......................................................................................................................................................................... 5 1. 2. 3. 4. 創薬開発における Proof of Concept (POC) ............................................................................... 8 1.1 創薬開発における POC とは ..........................................................................................................8 1.2 早期 POC 確保のための留意点(2.2 参照) ........................................................................9 早期 POC 確保のための開発戦略....................................................................................................... 10 2.1 バイオベンチャーの事業モデル................................................................................................. 10 2.2 早期 POC 確保のための開発戦略............................................................................................. 12 早期 POC 臨床試験を開始するための留意点 .............................................................................. 22 3.1 Chemical, Manufacturing and Control (CMC) ...................................................... 22 3.2 非臨床試験............................................................................................................................................. 25 3.3 開発対象となる適応症選択の重要性....................................................................................... 29 3.4 臨床試験.................................................................................................................................................. 30 3.4.1 開発戦略の重要性.................................................................................................................... 30 3.4.2 被験者の確保対策.................................................................................................................... 30 3.4.3 臨床試験の実施国の選定 ..................................................................................................... 31 3.4.4 国内か海外か?......................................................................................................................... 32 3.5 国内開発における Translational Research (医師の自主臨床研究) の活用 . 34 3.6 海外での早期 POC 臨床試験 ...................................................................................................... 37 3.7 日米欧における臨床試験の開始................................................................................................. 39 3.7.1 米国での治験.............................................................................................................................. 39 3.7.2 欧州での治験.............................................................................................................................. 43 3.7.3 日本での治験.............................................................................................................................. 45 3.7.4 アジア諸国での POC 臨床試験の可能性.................................................................... 46 まとめ................................................................................................................................................................. 47 参考文献等 ................................................................................................................................................................. 49 6 表目次 表 1 2006 年 医薬品売上メーカーランキング .............................................................................. 19 表 2 EU 諸国の治験申請審査に関わる状況........................................................................................ 44 図目次 図 1 ベンチャービジネスの死の谷 .......................................................................................................... 16 図 2 First in human trial 開始時に必要とされる非臨床試験(赤字の試験項目) .... 25 図 3 医師の自主臨床研究の成果の活用及び実施対応について ............................................... 35 7 1. 創 薬 開 発 に お け る Proof of Concept (POC) 1.1 創薬開発における POC とは 商品開発における”Proof of Concept(POC)”とは、その商品開発過程において、 その開発コンセプトの妥当性を傍証することと定義づけられる 2)。すなわち、仮説 設定から始まった商品開発が、その商品開発の真の目的となるニーズを満足させる ための実現性の傍証を固める段階にあることを意味する。POC を確保することは、 あらゆる商品開発に共通した開発プロセスにおける大きなマイルストンである。 創薬開発における POC とは、研究開発段階にある新薬候補について、その有効性 や安全性をヒトで探索し、その探索結果が創薬開発における開発確度を高めること につながる最も大きなマイルストンの一つであると同時に、創薬系バイオベンチャ ーにとっては製薬企業とのアライアンスの可能性を高め、更には企業成長の大きな 通過点である株式上場の機会をも創出する。 創薬開発における POC の確保は、バイオベンチャーや製薬企業にかかわらず、創 薬開発の開発確度を占う大きな道しるべである。 特に、資金の限られたベンチャー企業にとって、早期 POC の確保は企業存続の問 題にも直結しうる大きく高いハードルといっても過言ではないであろう。 8 1.2 早期 POC 確保のための留意点(2.2 参照) まず、創薬系バイオベンチャーにおける早期 POC 確保のための主な留意する点を 以下に示す。 新薬開発の真のゴールを明確に設定する ベンチャー企業経営としての一次ゴールを明確に設定する 経営者による開発担当者のスキルと性格の見極め ベンチャー企業経営の一次ゴール向けた資金運用 法令に基づいた“あるべき論”以外は、実行可能であることを意識す る(単なる経験値のみの“あるべき論”に対する警笛) 自社の弱みを正確に把握し、その弱い部分を外部の責任感のある信頼 のおけるパートナーに任せる(創業期) 製薬企業とのアライアンスを視野に入れ、欧米に信頼されるデータを 創出する(世界的なトップ 20 の製薬企業は主に欧米企業である) POC 臨床試験は、現時点では、欧米先行型がスピード、コスト、事 業戦略的にも有利である その他の留意点も数多くあると思われるが、上述した点は、事業開発の主軸を構築 するためには必要不可欠なものであると考える。簡潔に言うと、「目的を達成する ために、ニーズ対象に満足してもらえるような成果を、資金と人材を有効に活用し、 可能な限り早く創出する」ということに、常に敏感になるということに集約される のであろう。 ただし、先端医療領域については、技術進歩もさることながら原理原則となる法令 やガイダンスが成長過程にあるため、これらの変化にも柔軟に対応できる余裕をも った対応も必要である。最新の関連情報には、常に留意して開発を進めることが必 要であろう。 9 2. 早期 POC 確保のための開発戦略 POC 臨床試験は、真の目的の成功確度を高める傍証を固めるためのものであり、 この試験の結果そのものが真の目的と直結することは多くはない。症例数、実施医 療機関数が少なかったり、当該開発領域における主要評価項目の確保に長期間を要 するような場合は、副次的なエンドポイント等を活用したりする。開発スピードを 考慮すると、POC 臨床試験では、可能な限り短期に開発確度を確認するような試 験デザインが望まれる。POC 臨床試験に対する考え方は、創薬系バイオベンチャ ーや大手製薬企業の差はなく、共通理解であると考えて良い。 2.1 バイオベンチャーの事業モデル 創薬系バイオベンチャーにおける早期 POC 確保の重要性を多少述べてきたが、こ こではその重要性の理解を更に深めるために、バイオベンチャーの事業モデルにつ いて述べておきたい。 我が国では、米国と同様にゲノム解析やバイオテクノロジー系バイオベンチャーか ら、先端バイオテクノロジーによる創薬や、低侵襲型の手術機器等のバイオメディ カル系ベンチャーが主役になってきている。このような流れに同調し、受託解析ビ ジネスから創薬系ベンチャーに衣替えしてきたベンチャー企業も少なくない。言い 換えれば、確立しつつある自社の先端技術受託ビジネスを活用し、新薬や新医療機 器の開発を具現化する事業への転換である。三段跳びにたとえれば、“ステップ” の段階で、“ホップ”が成功したことにより、更なる飛躍に向けて“ステップ”し たいという事業戦略である。このような事業変換は、成功例を土台に、更なる発展 の感覚をイメージさせられ、投資家にはよりアピールできる事業モデルであった。 つまり、投資家に理解の得られるような事業モデルに変化することにより、更なる 資金を確保し、より強く企業が成長してきている。バイオベンチャーが継続的に発 展するためには、特に経営陣はあらゆる環境変化を敏感に感じ取り、柔軟に対応す ることが求められるのかもしれない。 一方、創薬系ベンチャーでは、がん、心血管系、糖尿病、精神神経系、眼科系又は 皮膚科系といった疾患や適応領域の専門化が顕著であるといえよう。また、最近で は、海外で上市されている医薬品や開発後期段階にある医薬品の権利を導入し、更 なる治験実施を含めたインキュベーション後、権利導出又は上市による利益確保を 10 目指す臨床開発特化型の創薬ベンチャーも誕生している。この事業モデルは、オリ ジナルシーズを基礎研究、製造、品質確保、非臨床開発、臨床開発と繋げる創薬ベ ンチャーに比べ、理論的には事業リスクが少なく、短期の収益確保が可能となる。 このような変化を伴う状況の中、我が国のバイオ投資環境は極めて悪く、米国に比 較し投資規模が小さいばかりか、最近では資金拠出を躊躇するベンチャーキャピタ ルが増え、資金調達がままならず事業縮小又は事業閉鎖に追い込まれているバイオ ベンチャーも出始めている。2003 年後半のアンジェス MG の上場を皮切りに、 2004 年末のタカラバイオの株式上場まで、国内バイオベンチャーの上場が続い たが、先行上場したバイオベンチャーが真の目的を目標どおりに達成することに苦 労し、株式上場後に期待どおりに一般投資家に応えることができない状況が続き、 その追い打ちをかけ株式上場基準が厳しくなり、短期の投資回収を目論むベンチャ ーキャピタルの投資熱も冷めてきてしまっている。我が国においては、長期的な視 点でバイオベンチャーを支える投資環境の整備も必要になろう。エンジェル税制の 導入等、国としても打開策は講じているが、根本的な解決には至っておらず、バイ オベンチャーの資金難は今なお継続している。真の成功例(医薬品の開発成功例) がきっかけとなり、このバイオ投資環境が少しずつ改善していくことに期待したい。 このような資金環境の中、創薬系バイオベンチャーには、これまで以上の“スピー ド感覚”が求められている。企業維持の経費と開発スピードを秤にかけ、時間優先 の開発戦略をとるために企業内のリストラクチュアリングは必至となるであろう。 また、短期での投資回収を期待するベンチャーキャピタルが少なくないことから、 バイオベンチャー側も短期でキャッシュ創出できるようなビジネスオプション等 も考えベンチャーキャピタルにアプローチすることも必要なのかもしれない。バイ オベンチャー同士又はライフサイエンス分野の別事業との M&A 等も真剣に考え なければならない時期に来ているのかもしれない。 11 2.2 早期 POC 確保のための開発戦略 これまで述べてきたように、創薬系バイオベンチャーにおいて、早期 POC 臨床試 験を実現できるか否かは、企業自体の存続にも影響しかねない。しかし、この臨床 試験を仕掛けたとしても良い結果が得られなければ、開発はその時点で混沌とする。 シーズのポテンシャルにより開発が結論付けられればまだ良いが、可能な限りヒュ ーマンエラーによる判断ミスは避けたいところである。 また、欧米のバイオベンチャーの新医療技術を応用した早期臨床試験の本数を見て も分かるように、一発必中で期待する結果を得るのは困難を極めている。したがっ て、満を持して臨床試験を仕掛ける姿勢よりも、スピードを重視し、変化しながら 本丸となる POC 確保を目指す戦略のほうが現実であると考える。新薬開発におい ては、満を持したところで、開発の成功確度を急激に上昇させる確率は高くないと 考えられることから、いち早くヒトでのレスポンスを感じ取ることが、創薬系バイ オベンチャーに優先されることは自明であろう。 1.2 項で、創薬系バイオベンチャーにおける早期 POC 確保のための主な留意点に ついて述べたが、そこに列挙した留意点は、当該開発領域における最新の確度高い 情 報 、 早 期 臨 床 試 験 が 可 能 な 国 の 薬 事 事 情 、 FDA ( Food and Drug Administration; 米国食品医薬品局)や EMEA(European Medicines Agency; 欧州医薬品庁)に対する研究開発データの acceptability 及び当該開発企業の開発 能力の冷静な分析のもと、当該開発品目の真のエンドポイントと当該企業の一次ゴ ールを見定めるために必要な項目である。以下に、その留意点について簡単に解説 する。 1) 新薬開発の真のゴールを明確に設定する この留意点には、将来予想も含まれるため、これまでの同種開発領域の開発経 験やある種の“勘”も求められる最も深く難しい課題である。開発領域にもよ るが、一般的に創薬系バイオベンチャー独自で後期第2相臨床試験以降を単独 で行うことは、資金的事情からも非常に厳しい。したがって、POC 臨床試験 又は前期第2相臨床試験の成果をもって、製薬企業とのアライアンスを成立さ せ、その後の臨床試験をライセンス先の製薬企業で行うか、共同で行うことが 現実的である。事実、欧米のバイオベンチャーは、このようなスキームを実現 12 し、成長発展してきている。 バイオベンチャー側としては、製薬企業が興味を持つような適応症やマーケッ トポテンシャルを常に意識し、開発を進める必要がある。 Unmet Medical Needs(未だ有効な治療法のない医療ニーズ)医薬品で、比 較的大きなマーケットの開発領域については、大手製薬企業が鎬を削って開発 競争を繰り広げている。がん治療の領域は、まさにこのフィールドといって良 いであろう。既存療法が存在する領域であれば、どのような治療ポジションを 確保するかの仮説をヒストリカルデータや治療領域の専門家の意見も参考に しながら理論的に構築し、開発戦略を策定することになる。この場合、特に留 意しなければならないことは、欧米と我が国の治療環境が異なる治療領域につ いては、多く場合、先に欧米仕様を前提に開発戦略を構築することが余儀なく されることである。現時点では、国内製薬企業の国内バイオベンチャーに対す る評価は高いというところまで来ておらず、アライアンス先に国内製薬企業の みを早期に設定することは事業的には大きなリスクを背負うと思われる。また、 既存薬が存在することは、第3相臨床試験での比較対象薬が存在することを意 味し、開発前例も存在することから、その開発データを規制当局が保持してい る。この点に注目すると、開発当初から真のゴールとなる適応症全般をターゲ ットにした場合、既存薬の承認前例を参考に非臨床試験パッケージを構築しな ければならない可能性も高まる。これは、臨床試験に入るまでに時間がかかる 大きなハードルとなる。 したがって、多くのバイオベンチャーでは、真のゴールは理想的なマーケット ポテンシャルがある適応症全般だが、まずは開発スピードをつけるために、重 症度が高い患者等に開発のターゲットポピュレーション等を絞り込み、リスク ベネフィットを高く勘案することができる Unmet Medical Needs 領域を創 出する開発戦略を策定する。Unmet Medical Needs 領域の開発は、確立し た根本治療法が存在しないことから、患者及び医療現場のニーズも高く、早期 開発が望まれることから、非臨床試験のスモールパッケージ化も不可能ではな い。また、医師の開発意欲も高まるため、開発も強く早く進む可能性が高くな る。 開発スピードに関わる大きなリスク因子としては、ターゲットポピュレーショ ンを絞り込んだことにより、当該臨床試験の対象患者が少なくなり、症例登録 13 に時間がかかることかもしれない。予定期間内に臨床試験を終了させるための 実施医療機関のフィージビリティー調査は確度高く行いたいところである。 「新薬開発の真のゴールの設定」は、様々な開発ストーリー(開発戦略)オプ ションの根幹となることから、開発戦略策定時の最も重要な留意点となる。 2) ベンチャー企業としての一次ゴールを明確に設定する ベンチャー企業としての一次ゴールを経営面から考えると、多くの場合、その 一次ゴールは株式上場となるであろう。創薬開発には莫大な資金が必要になる ことから、創薬系バイオベンチャーにとっては市場からの資金調達は必至であ る。更に、多くの場合、ベンチャーキャピタルをはじめ、多くの投資家からの 資金調達を繰り返し行っているため、投資家のゴールである株式上場は通過し なければならない大きなマイルストンとなる。創薬という観点からは、株式上 場は通過点に過ぎないが、企業経営的には一次ゴールといってもよいであろう。 3) 経営者による開発担当者のスキルと性格の見極め 創薬開発の成功の秘訣は、優秀な人材の確保といっても過言ではない。創業初 期は、医薬品製造のスペシャリストの存在が必須である。特に、バイオ医薬品 の場合、いわゆる CMC(Chemical, Manufacturing and Control)といわ れる製造及び当該開発品の品質確保分野への対応が重要なウエイトを占める。 開発の早期成功の可否の 6~7 割は、この CMC 対応で決まるといっても良い かもしれない。 また、2.2 項 1)で述べた真のゴールを設定し、開発ストーリー及び事業計画 を構築するためには、当該開発疾患領域の開発経験をもつ臨床開発の専門家、 特に開発企画のスキルが高い人材を確保することが理想的であろう。バイオベ ンチャーには、いわゆる臨床試験モニターのスペシャリストは当面必要なく (外部支援で手当可能)、企画立案能力に優れた開発の専門家が存在すること が望ましい。しかし、このような人材は、大手製薬企業でも貴重な存在であり、 なかなか転職市場には出てこないのが現状である。このような“優秀な臨床開 発のスペシャリスト”不足は、我が国の創薬系バイオベンチャーの大きな課題 でもある。 欧米では、製薬開発能力の高い、開発疾患領域に強い MD(Medical Doctor; 医師)がベンチャー企業に参加しているケースがほとんどであり、我が国の抱 14 えるような人材難の問題はあまり聞かない。我が国も MD の活躍領域が広が り、欧米と同じような環境になる時代が来ることを期待したい。 非臨床試験については、米国チャールスリバー社やコバンス社のような大規模 な CRO(Contract Research Organization; 開発業務受託機関)であれば、 各種バックグラウンド情報も豊富で、同種の開発品目の試験経験もある可能性 が高く、デリケートな試験も比較的安心して任せられる可能性が高い。また、 コンサルティング能力も高く、ベンチャー企業にとって大きな味方になるであ ろう。可能であれば、創薬系バイオベンチャー内に非臨床試験担当者もほしい ところだが、良いコンサルタントにめぐり会えば、外部対応も不可能ではない。 なお、バイオベンチャーに必須なウエット研究を行う研究員が必要なことは言 うまでもない。 このように、早期に POC 臨床試験を行うには、上述のような企画能力に長け た優秀な人材を採用することが望ましいが、この優秀という言葉の中には性格 やベンチャースピリッツというものも含まれてくることを忘れてはならない。 ベンチャーにとって、積極果敢に攻めるマインドは必須であり、いくら優秀で あってもこのようなマインドが伴わなければ、常に過剰にリスク優先で物事を 判断してしまい、結局は物事が前に進まなくなってしまう。開発の主要担当者 のスキルとマインドのバランスを経営陣がしっかりと把握し、自社の開発面に おける弱点を見極めることは大きな課題である。また、ベンチャーの開発現場 に必要なことは、立派な経歴よりも実行力であることが少なくない。時には、 経営陣自身が現場介入し、経営陣の目で開発状況を確認するような厳しい対応 も必要であると考える。 4) ベンチャー企業経営の一次ゴールに向けた資金運用 創薬系バイオベンチャーは資金調達を繰り返して、図 1 に示すようなベンチ ャービジネスの死の谷を凌いでいく。 15 図 1 ベンチャービジネスの死の谷 2.2 項 2)で述べたとおり、ベンチャー企業の経営面での一次ゴールは株式上 場であり、まずはこの株式上場を達成すべく、ベンチャーは資金調達を繰り返 す。資金調達を繰り返すには、資金調達ごとの成果マイルストンを明確に設定 し、その妥当性をベンチャーキャピタリスト等に理解させ、更なる資金提供を 求めることになる。 開発が進むにつれ、人員も増え、株式上場に向けて管理部門の強化も求められ ることから、開発面以外の経営管理的な維持固定費の増加も顕著になる。大手 製薬企業の場合、開発の遅れに対する人件費等の維持固定費は、企業経営の中 で吸収されるが、ベンチャー企業ではこの遅れが、直接、死活問題につながる 可能性は低くはない。著者は、社員一人に対し、企業の維持経費(人件費、オ フィス賃貸料や活動費等を含む)が、平均年間 1,000 万円を下らないことを 経験している。したがって、例えば 20 名で構成されるベンチャー企業(役員 を含む)の年間維持費は、少なくとも 2 億円はかかるわけである。このベン チャー企業で、開発計画が 3 カ月遅れた場合、単純に考えれば 5,000 万円を 損失したことになる。スピードが如何に大切になるかが理解できる。 このように開発の進展や株式上場による人員増及び施設設備の増大等、企業成 長に伴う維持固定費の増大と、資金調達のマイルストンとなる開発成果の実行 確度は、時期的な面も含め慎重に検討し、経営上のクライシスマネジメントを 16 行う必要がある。場合によっては、人員削減に踏み切る決断を余儀なくされる 場合もあるであろう。経営資源の限られたバイオベンチャーの場合、予防的に 経営リスクに対応する必要がある。予防的に対応しなければ、経営破たんのリ スクが増大する可能性も否定できない。 5) 法令に基づいた“あるべき論”以外は、実行可能であることを意識する(単な る経験値のみの“あるべき論”に対する警笛) 法令遵守は、企業経営も医薬品開発もまったくその位置づけは同様で、原理原 則となる法令を無視した事業展開及び開発展開は成立しない。 しかし、医薬品開発においては、“経験則によるあるべき論”というものが存 在する。例えば、細胞・組織医薬品等の非臨床試験ついての明確なコンセンサ スは、現時点では存在しておらず、非臨床試験 1, 2 本で臨床試験に移行して いる例も存在する。このような環境の中で、低分子医薬品開発経験における“あ るべき論”を展開してしまったらどうなるであろうか。少々極端な例であるが、 先端的な医療技術を用いた医薬品開発については、その考え方も成長過程の場 合があり、従来の常識が通用しない場合もある。特に、バイオ医薬品の場合、 ケースバイケースで判断することが少なくない。 したがって、創薬系バイオベンチャーの場合、ケースバイケースでの思考スタ ンスを常に保持し、法令遵守に抵触すること以外はまずは実行可能と考え、あ らゆる早期開発オプションを検討したほうが良いと思われる。もちろん、この ような検討の中で、“倫理”や“道義”面に不安要素があるものは選択するべ きではない。このような不安要素を抱えて開発を進めることは、医薬品開発の 長い開発過程で、必ずいつか問題提起されるものである。特に、人の健康に直 接寄与する業務形態の創薬系バイオベンチャーにとって、“倫理”や“道義” 面での社会的な批判は企業生命にも影響しかねないことから十分な留意が必 要である。 一方で、強い企業理念のもと、法令遵守を原理原則として、ケースバイケース で積極的にあらゆる角度から物事を評価する習慣は身につけていきたいもの である。 6) 自社の弱みを正確に把握し、その弱い部分を外部の責任感のある信頼のおける パートナーに任せる(創業期) 2.2 項 3)でも述べたが、まずは経営陣による自社の開発力の見極めが大切で 17 ある。自社の開発面での弱みを正確に把握し、その弱い部分を責任感のある信 頼のおける外部パートナーに任せる勇気も必要である。 弱点が見極められているベンチャーほど、考え方の軸がぶれず、しっかりとし た強さを持っている。しかし、この弱みは、企業成長とともに解消していくも のと思われ、創薬系バイオベンチャーの創業期に特有な問題であるのかもしれ ない。資金的に一番厳しい創業期に、速やかに的確な対応ができるベンチャー がまず一歩リードしていくのであろう。 7) 製薬企業とのアライアンスを視野に入れ、欧米に信頼されるデータを創出する (世界的なトップ 20 の製薬企業は主に欧米企業である) まず初めに 2006 年の製薬企業の売上ランキングの上位 20 社を以下に示す。 18 表 1 順位 2006 年 医薬品売上メーカーランキング メーカー名 本社所属国 売上高(百万ドル) 1 ファイザー アメリカ 45,083 2 グラクソ・スミスクライン イギリス 39,335 3 サノフィ・アベンティス フランス 37,461 4 ノバルティス スイス 29,491 5 ロシュ スイス 27,318 6 アストラゼネカ イギリス 25,741 7 ジョンソンエンドジョンソン アメリカ 23,267 8 メルク&Co. アメリカ 22,636 9 ワイス アメリカ 16,884 10 イーライ・リリー アメリカ 14,816 11 アムジェン アメリカ 14,268 12 ブリストルマイヤーズスクイブ アメリカ 13,861 13 アボット・ラボラトリーズ アメリカ 12,395 14 ベーリンガー・インゲルハイム ドイツ 11,637 15 バイエル・シェーリングファーマ ドイツ 9,873 日本 9,613 17 ジェネンテック アメリカ 9,284 18 シェリング・プラウ* アメリカ 8,561 イスラエル 8,408 日本 7,717 16 武田薬品工業 19 テバ製薬工業 20 アステラス製薬 データは、ユートブレーン LLC 合同会社(http://www.utobrain.co.jp/)の発表に基づく。 *シェリング・プラウの売上にオルガノンは含まない。シェリング・プラウ+オルガノンの場合は 14 位。 19 当該 20 社のうち、18 社が外資系企業で、国内企業は武田薬品工業及びアス テラス製薬のみの2社である。資本力及び開発力が大きい製薬企業は海外に多 いことから、製薬企業とのアライアンス戦略もまずは海外中心で行くことが、 確率論的な考え方からは妥当な選択であろう。 したがって、当該アライアンス確率を向上させるためにも、海外製薬企業に魅 力のある信頼される臨床試験データの創出が望まれる。最近の大手国内製薬企 業の売上も、その多くの部分が海外に依存しているため、海外マーケットを重 視した開発戦略であれば、結果的に国内製薬企業も興味を持つことになると思 われる。アライアンス戦略のみを考えると、POC 臨床試験は白色系人種のデ ータのほうが、好ましいのかもしれない。 しかし、最近は、アジア諸国において患者が多い肝疾患(HBV、HCV、肝が ん等)やある種の感染症(HIV、H5N1 influenza virus 等)治療薬の開発に おいては、真のゴールを強く意識し、臨床試験のコストやスピード面も重視し たアジア諸国中心の開発に注目している企業も出てきている。世界人口の大半 を占めるアジア地域の医薬品マーケットは、これから急成長することが予測さ れているため、これからのアジア地域は注目すべき医薬品開発フィールドにな るであろう。アジア諸国の早期臨床試験の品質は、未だ議論の余地は残される ところであるが、過去の我が国もそうであったように、比較的近い将来に worldwide standard の臨床試験の品質となることは間違いないであろう。 8) POC 臨床試験は、現時点では、欧米先行型がスピード、コスト、事業戦略的 にも有利である 2.2 項 7)で製薬企業とのアライアンスを視野に入れ、欧米に信頼されるデー タを創出することの重要性を述べたが、このようなことからも海外データ(白 色系人種データ)の取得を第一に考えることが妥当であろう。国内データが完 全に不利になるということではないが、臨床試験への被験者登録のスピードや 臨床試験の実施コストを考えると、時間的にも費用的にも海外開発が優先され る。特に、国内治験では、国策となりうる特殊な疾患を別にすれば、臨床試験 への被験者登録のスピードは海外に比較して遅く、その分コストもかかる。 第 3 回の「ヒト POC に関する勉強会」で、アステラス製薬臨床管理部の津田 課長にご講演いただいたが、国内 CRO にモニタリングを委託するときの経費 は、モニターの必要経費を含め 250 万円/モニター/1 箇月程度であるという お話であった。更に、日本の GCP が非常に厳格な対応を求めているため、担 20 当施設も 3 施設/モニター程度になるとのことであった。著者は、モニター1 名のみを確保するために、国内大手 CRO へのモニタリング依頼を検討した際 に、CRO 側からプロジェクトリーダーやモニタリングリーダーの採用も求め られる場合を経験している。モニターを 1 名確保したいがために、3 名体制 でのモニタリング体制の委託が余儀なくされた場合、CRO へのモニタリング 委託経費のみで、少なくとも 1 箇月に 500 万円程度はかかる計算になる。こ れを年間経費に換算すると 6,000 万円という数字がはじき出される。資金状 況の厳しいベンチャー企業にとって、臨床試験の 1 業務のみにこの費用をか けるのはかなり勇気ある決断を迫られる。 また、通常、モニタリング委託は治験ごと、症例ごとでの費用算定にはならず 拘束期間での費用算定となるため、2 年、3 年と臨床試験の期間が延長される につれ、費用も上昇してしまう。よく海外との治験費用の比較がされ、我が国 の費用の高さが述べられるが、同一期間内での臨床試験業務の単価を考えると 試験総額ほどの大きな格差はないように感じられる。我が国では、海外と比較 して、一般的に臨床試験の実施に時間がかかると言われており、その長期化し てしまった期間分が国内臨床試験のハイコストを誘導しているのかもしれな い。 我が国の創薬系バイオベンチャー企業における国内開発優先戦略は、臨床試験 確保という意味からも、しっかりとした資金の後ろ盾があって、安心して進め られるのであろう。 なお、本項で述べてきた国内での臨床試験の費用については、あくまでも一般 的な国内状況を述べたものであり、ベンチャー企業に対し、可能な限り費用を 抑えた優しい契約オプション提示をしている CRO 企業も存在していることは 申し添えておきたい。 21 3. 早期 POC 臨床試験を開始するための留意点 本章から、早期 POC 臨床試験を開始するための留意点の各論に入りたい。紙面の 都合から概略を示すにとどまるが、特に、ここでは創薬系バイオベンチャーの生命 線ともいえるバイオ医薬品の開発について述べたい。低分子医薬については、既に 多くの開発実績があり、我が国でもその研究開発にはある一定の考え方が存在して いることから、本章ではバイオ医薬品の早期 POC 臨床試験を開始するための留意 点について概説する。 3.1 Chemical, Manufacturing and Control (CMC) バイオ医薬品の CMC は、開発の根幹を担うものであり、特に、最終製剤の品質は 安全性に直結することから、その重要性の比重は臨床試験を実施することよりもは るかに重い。開発初期段階では、GMP(Good Manufacturing Practice; 医薬 品の製造に関する基準)に準拠した製造は求められるものの、一般的にコマーシャ ルスケールでの製造を要求される第 3 相臨床試験前より、GMP に対する考え方の フレキシビリティーは高く、規制当局とのコンセンサスのもと対応できる。我が国 では、治験薬 GMP という基準が存在し、海外では開発領域ごとに関連ガイダンス が出されたりもしている。例えば、技術革新が激しい領域においては、米国 FDA は規制当局の審査員及び開発企業の双方に対し共通理解を求めるべく、以下のよう なガイダンスも発行している。 ・ Draft Guidance for FDA Review Staff and Sponsors: Content and Review of Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs) - 11/8/2004 規制当局の審査員及び開発企業の双方に経験値や十分な知識が蓄積されていない 状況で、先端技術に対する道しるべを作っている。新しい医療技術を米国国家とし て、しっかりと支え、強く育てる積極的な産官連携の良いモデルである。 バイオ医薬品の抱える大きな問題として、開発ステージに応じた製造スケールの拡 大がある。通常、開発スピードやコストの関係もあり、開発当初から何トンもの培 養槽を用いた製造は行わず、first in human trial 開始時には、いわゆる pilot scale レベルでの製造が行われる。この pilot scale 製造(数百リッターレベルの培養槽 22 での製造が多い)時点で、ベンチャー企業からの CMO(Contract Manufacturing Organization; 製造業務受託機関)への製造技術移転が完了する。多くの場合、 この技術移転時に、原薬及び最終製剤の暫定規格、規格設定のための試験方法の確 立並びに安定性試験が行われる。多くの創薬系バイオベンチャーの場合、この CMO での製造原薬で、治験申請のための安定性試験及び非臨床試験データを収集し、最 終製剤の安定性試験成績を添えて治験申請に臨む。原薬製造と製剤製造の場所が異 なる場合もある。その後、臨床試験の規模の拡大とともに、製造場所や製造工程の 変更を重ねて当該バイオ医薬品は開発ステージとともに育って行く。しかし、バイ オ医薬品の製造場所や製造工程の変更は、最終製剤の品質プロファイルを変更して しまうリスクがあり、スケールアップにもこのリスクが存在する。原体の3次構造、 最終製剤の夾雑プロファイルや生物活性が大きく変わってしまうケースがある。も し、旧製法と新製法のバイオ医薬品のプロファイルがまったく異なるものになって しまった場合は、最悪の場合、一からの開発のやり直しを余儀なくされることも想 定される。 創薬系バイオベンチャーの CMC 担当者は、このようなリスクを事前に想定しなが ら、スケールアップ戦略を考えることが望まれる。 筆者が経験した多くの場合、旧製造場所・旧製法と新製造場所・新製法での各々の 最終製剤におけるコンパラビリティー(同質性)を品質データで証明することで対 応してきた。多くのケースでは、品質向上のための製法改善となるため、安全性向 上のストーリーは容易に作りやすい、問題は生物活性のコンパラビリティーである。 生物活性の分析は、通常、標準物質との比較検討を行い、この標準物質に対し、例 えば 70%以上の活性を維持していれば規格範囲としたりする。一般的に、生物活 性の分析データのばらつきは大きいと言われている。このように大きなばらつきを もつ開発途上の分析法しか確立していないときのコンパラビリティー検討にはい つも苦労する。また、規格値によるコンパラビリティー検討には注意を要する。こ の場合、比較データがお互い単純に規格範囲にあればよいというわけではなく、そ の規格範囲内でのばらつき傾向等にも留意しなければならない。したがって、この ようなコンパラビリティー検討には、通常、少なくとも3ロット程度の品質データ は必要とされるであろう。早めに規制当局との相談を行っておくことが無難であろ う。 結論としては、開発ステージのどの時期で、スケールアップを行うのかを製薬企業 とのアライアンス戦略も含めた形で想定し、社内の経営戦略も加味して事前に検討 23 しておくことが望ましいと考える。バイオ医薬品の CMC には、開発を左右するよ うなデリケートな要素も秘められていることを常に意識しておきたい。 基本的には、ICH Q5 シリーズガイダンス 3)及び Q6B ガイダンス 4)については、 十分に把握しておくことが必要である。ただし、遺伝子治療用医薬品等の一部のバ イオ医薬品については、これらのガイダンスの対象外となっているものも存在する ので留意されたい。 24 3.2 非臨床試験 新規医薬品候補物質を初めてヒトに臨床応用する際には、その薬効や安全性を確認 する非臨床試験を行わなければならないことは言うまでもない。参考までに、ICH M3 ガイダンス 5)において、first in human trial 開始時に必要とされる非臨床試 験を、図 2 に記載されている赤字の試験項目で示す。 図 2 First in human trial 開始時に必要とされる非臨床試験(赤字の試験項目) 25 参考までに、ICH M3 ガイドラインに記載されている初めてヒトを対象とした臨床 試験を行う前に必要とされる非臨床試験安全性試験に対する考え方の概略を以下 に示す。 1) 単回投与毒性試験 初めてヒトを対象とした試験を行う前に、2種の動物を用いた単回投与毒性 試験結果が必須である。 2) 反復投与毒性試験 原則として臨床試験での投与期間を超える期間の反復投与毒性試験の結果に 基づいて臨床試験の実施を検討する。 3) 遺伝毒性試験 初めてヒトを対象とした試験を行う前に、in vitro の変異原性及び染色体異 常試験を行う。標準的な組み合わせの遺伝毒性試験は、第2相試験開始まで に行う。 4) 安全性薬理試験 初めてヒトを対象とした試験を行う前に、呼吸・循環器系及び中枢神経系へ の有害作用など生命に関わる影響の評価を行う。 5) トキシコキネティクス及び薬物動態試験 毒性試験と関連した暴露データは、初めてヒトを対象とした試験を行う前に 必要である。これ以外の薬物動態試験結果は、通常、第Ⅰ相試験が終了する までには得ておく。 6) がん原性試験 特別の理由が無ければ、臨床試験実施前に終了していなくともよい。 7) 生殖発生毒性試験 雄受胎能試験は第3相試験の前でもよい。妊娠の可能性のない女性での試験 は、生殖発生毒性試験結果が得られる前に行ってもよい。妊娠可能な女性を 対象とする際に必要な生殖発生毒性試験の範囲については、三極の要求は統 一されていない。我が国では雌受胎能試験と胚/胎児発生試験結果が必要とさ れている。 8) 小児での臨床試験 小児試験の実施の際には、通常、事前に成人での情報を入手しておく。また、 適切な種類の反復投与毒性試験と生殖発生毒性試験、及び標準的な組み合わ せの遺伝毒性試験も事前に行っておく。小児試験実施の際の幼若動物を用い 26 た試験の必要性については、個々の事例に基づき判断する。がん原性試験の 必要性についても、投与期間や他の情報をもとに判断する。 上述のような非臨床試験に関する基本的なスタンスに加え、バイオ医薬品の非臨床 試験における安全性評価については、ICH S66)ガイダンスに取りまとめられてい る。このガイダンスの一般原則は以下のように述べられている。 <ICH S6 ガイダンスに記載されている一般原則> 非臨床安全性試験の目的は、臨床試験実施前のみならず臨床開発の全段階を通じ て、医薬品の薬理作用及び毒性作用を明らかにすることである。In vitro 及び in vivo 試験のいずれをも実施することで、これらの特性が明らかとなる。臨床的に 広く使用されている医薬品と構造的及び薬理学的に同等であるバイオ医薬品の場 合、毒性試験を簡略化してもよいことがある。非臨床安全性試験においては、以 下の点について考慮しなければならない。 適切な動物種の選択 齢 生理的状態 投与量、投与経路、投与方法等を含めた投与計画 使用条件下での被験物質の安定性 毒性試験は、「医薬品の安全性試験の実施に関する基準(以下、GLP という)」 に適合して実施されることが求められている。しかし、バイオ医薬品で必要とさ れることの多い特殊な試験系の中には、完全には GLP 適合で実施することができ ないものがある。このような場合には、GLP に適合していない部分を明確にし、 安全性評価全体に対する重要性について評価しなければならない。GLP に完全に は適合していない試験でも、得られるデータが臨床試験の実施や製造承認申請の ために用いてはならないことを必ずしも意味しない場合がある。 バイオ医薬品ではその特徴的かつ多様な構造並びに種特異性、免疫原性及び予想 外の多形質発現活性の生物学的性質のため、医薬品の従来の毒性試験法が適切で はない場合がある。 27 また、バイオ医薬品の特性から、動物での安全性検討が確度高くヒトでの安全性を 外挿するとは言い難いようなケースにも遭遇するようになってきた。このような場 合の非臨床試験パッケージは、ケースバイケースで判断されることになり、事前に 規制当局としっかりコンセンサスを取っておくことが開発をスムースに進めるた めには必要不可欠であると考える。 また、遺伝子治療用医薬品や細胞・組織利用医薬品等では、ICH S6 ガイダン スが適用されず、このような高度先端医療領域の医薬品開発においては、規制 当局との綿密なコミュニケーションを余儀なくされるであろう。我が国におい ては、当該開発領域に確認申請 7)という制度があるが、この審査で規制当局と 品質と安全性の深い議論を行うことになる。 一方で、バイオ医薬品の特性や開発戦略によっては、必要最小限の非臨床パッ ケージで、初めてのヒトでの臨床応用を可能にできることもある。 28 3.3 開発対象となる適応症選択の重要性 新規医薬品を開発するに当たっては、まず開発目標となる適応疾患を定めるのが一 般的である。2.2 項 1)でも述べたように、創薬系バイオベンチャーとしては、製 薬企業とのアライアンスも視野に入れ、製薬企業が興味を持つような適応症やマー ケットポテンシャルを考慮した開発戦略を構築する必要がある。したがって、まず は Unmet Medical Needs(未だ有効な治療法のない医療ニーズ)医薬品でマー ケットが比較的大きい開発領域への開発展開を探ることになる。この Unmet Medical Needs については、いわゆる既存薬が存在しないということのみではな く、既存薬はあるがその価格が非常に高い又は使用制限があり入院治療や高額医療 を余儀なくされる等の医療経済的貢献度の高い医薬品群も対象になり得ると考え る。全世界的に国民医療費が国家の財政を圧迫し、どの国も安価で安全で効果のあ る薬を望んでいる。 Unmet Medical Needs 医薬品や国策として開発を急がれる医薬品(Pandemic influenza virus vaccine 等)については、その開発が急務とされ、社会的にも一 日も早く有効な対処方法の機会を得たいことから、リスクベネフィット評価が積極 的に行われ、非臨床試験パッケージもコンパクトに抑えられる場合も多い。また、 リスクベネフィットを勘案し、非臨床試験からでは排除しきれないリスクを臨床試 験で確認しながら進めることが認められるケースもあり、ケースバイケースの判断 は積極的に拡大する。 開発スピードを重視し、製薬企業とのアライアンスを想定した早期開発可能な臨床 試験の適応の選択が、POC 臨床試験までの非臨床試験を最小化する可能性もある ため、この臨床試験における適応を事業戦略・開発戦略とともに明確に定め、効率 的な資金運用を検討することにも留意したい。 29 3.4 臨床試験 ここでは、POC 臨床試験又は first in human trial について概説したい。創薬系 バイオベンチャーの場合、製薬企業とのアライアンスの確度を上昇させ、更に株式 上場のチャンスを創出するまでは、その資金的事情から、当該臨床試験の実施はよ り早く完結させ、より安価で対応したい。2.2 項 7)でも述べたように、アライア ンスチャンスは外資系製薬企業のほうが、確率論的には高い可能性があることから、 一般的には海外での臨床試験の実施をまず考えることになるであろう。以下に、こ れまでの「ヒト POC に関する勉強会」での議論も踏まえ、早期に臨床試験を実施 するための留意点を含め、早期 POC 確保のための留意点について述べたい。 3.4.1 開発戦略の重要性 欧米の創薬系バイオベンチャーの開発戦略は、一般的に Unmet Medical Needs 領域を創出し、開発にスピード力をつけ、効率的に短期間での POC 臨床試験を完 結する戦略を策定する。資金力が十分でない創薬系バイオベンチャーにとっては、 セオリー的な開発戦略になっている。 この戦略の一番の問題は、Unmet Medical Needs 領域を創出することにより、 対象患者が絞られ、臨床試験への症例登録が長期化することである。創薬系バイオ ベンチャー企業が POC 臨床試験を行う際に最も大切な確認事項の一つは、実施医 療機関の綿密なフィージビリティー調査を行い、治験申請後、速やかに臨床試験が 開始できる地域を検討し、選択することに尽きる。 対象疾患の分布によっては、アジア地域が優先的に選択される場合もあることにも 十分に留意されたい。 3.4.2 被験者の確保対策 筆者を含め、我が国の創薬系バイオベンチャー企業の臨床開発担当者に最も欠けて いると思われることが、被験者確保のための実施医療機関のフィージビリティー調 査かもしれない。信憑性の高い対象患者の数値データから、臨床試験の対象患者を 想定し、スクリーニング脱落率及び同意取得率を乗じ、年間登録推定患者数を割り 出す必要がある。著者は、これまで 20 年ほど臨床開発を経験してきているが、国 内の医師から意見聴取により得た試験適格年間患者数の情報は、その 1/3 程度が 30 実際の現実的な年間登録患者数になるようである。調査する開発企業側が医師に的 確に判断する情報を十分に与えていないことも、このようなギャップを生んでしま う要因になるのかもしれない。 創薬系バイオベンチャーの臨床試験責任者は、最終的には、実施医療機関の担当医 師や医療機関関係者と直接話し、事前に入手した各種の情報についても自身で現場 レベルでの確認を行うことが重要である。CRO からのレポートの書面評価だけで はなく、最終的には、臨床開発責任者が自ら実施医療機関に足を運び、臨床試験の フィージビリティー確認を行う必要があると思われる。 また、最近、著者が経験したことであるが、いわゆる“Big pharma”と呼ばれる ような製薬企業が、創薬系バイオベンチャーと同領域でほぼ同時並行でいくつも開 発を行っている時には、特に海外開発においては更なる注意を必要とする。“Big pharma”はその資金力と人海戦術で、良い実施医療機関を根こそぎ確保してしま い、ベンチャーが後追いするようになってしまうと、予定していた実施医療機関へ の治験依頼ができなくなってしまうことがある。海外では、治験を実施する医師と 製薬企業との直接契約になるため、製薬企業が望む期間内に望む症例数が確保でき ないと治験担当医師が判断した場合は、治験契約には至らない。患者が多く集まり、 良い医師がいる病院は根こそぎ“Big pharma”に取られ、ベンチャーはその隙間 を縫って開発を進めることになる。このようなことになると、開発スピードは上が らず、苦戦を強いられることになる。 同領域の他社の開発状況は常に意識し、場合によっては、治験実施医療機関レベル での情報を収集したいところである。 3.4.3 臨床試験の実施国の選定 臨床試験の実施国選定の際の検討項目を以下に示す。試験期間、開発コスト又は製 薬企業とのアライアンスに影響を及ぼす検討項目となる。 治験申請後、first patient in までの期間が短い 実施国のおける対象患者数及び実施医療機関における対象患者数 臨床試験実施地域 試験費用 臨床試験のマネジメント 31 海外からの治験誘致を経済活性政策に位置づけているような国では、規制当局によ る治験申請審査期間も比較的短く、積極的なコミュニケーションも可能となる場合 が多い。 3.4.4 国内か海外か? 製薬企業とのアライアンス確率の向上、開発コスト・スピードを考えると海外先行 型が優先されるであろう。 第2回の「ヒト POC に関する勉強会」において、参加企業9社及び講師として招 聘した東大の小野先生に対し、本勉強会で国内又は海外どちらの開発を先行させる かのアンケートを取ったが、1社を除きすべての方が欧米先行主導との回答であっ た。欧米以外(日本を除く)での海外実施の回答はなかった。臨床試験データの世 界標準としての信頼度、開発コスト・スピードがその主たる理由であった。 しかし、臨床試験のマネジメントについて、CRO 依存型かコンサルタント依存型 かということについては意見が分かれた。開発領域に強い CRO、規制当局とのコ ミュニケーション能力の高いコンサルタント、複数国を統一基準で管理対応できる CRO 等、それぞれ臨床試験のマネジメントにも特徴があるため、自社プロジェク トのコンセプトに合ったマネジメントを選択することが望ましいと思われる。また、 一般的に我が国のバイオベンチャー企業は、海外 CRO にとって、いわゆる“良い お客さん”になりがちなので、しかるべきルート(強い紹介ルート)で導入交渉し たり、海外 CRO が提案してくるサポートオプションをしっかりと評価したりする ことが必要となるであろう。我が国の多くのバイオベンチャー企業は、海外 CRO の提案をすべて受け入れてしまいがちなので、いわゆる“良いお客さん”になって しまう。 臨床試験の CRO への委託は、少し条件を変えるだけでも、数千万円の違いが出る ことがあることから、慎重に検討したいところである。 また、海外 CRO への“丸投げ”は、開発リスクを高めることが少なくないので注 意を要する。海外 CRO にとって、我が国のバイオベンチャーの仕事は、金額的に は大きなものではなく、常にその業務優先順位は下部に位置してしまう。この優先 順位を少しでも上昇させるには、相手が緊張感を維持するような環境を作り、場合 によっては海外の医師や研究者と一緒に海外 CRO を訪れプレッシャーを与えるこ 32 とも必要であるかもしれない。とにかく、はじめが肝心である。 著者がよく行うことは、大手外資製薬の開発部長クラスから紹介を取り付けたり、 著者自身が ICH 関係者のため、その開発地域での審査関係者との近さをアピール したりする。このような導入により、CRO の緊張感を高めている。また、その後 の継続的な経過観察訪問も大変重要である。現在は、TV 会議等で模擬的な face-to-face の環境はつくれるが、これはバーチャル環境にすぎず、緊張感もや や弱いことから、やはり自身で足を運んで相手と話したり、時には懇親の意味で食 事をしたりすることも重要であると考える。臨床試験が1個月延長することを考え れば、数回の海外渡航費など安いものとなる。また、CRO は開発企業にとって良 きパートナーでなければならないことから、単なる強制的な管理ではなく、CRO がモチベーション高く仕事に携わることができるような関係を構築することも重 要なことであると言えよう。 臨床試験においては、できる限り、物事は予防的にとらえ、事後的な対応は避けた い。また、予防的な活動に費用を積極的に使うことを考えていきたい。我が国では、 予防的に対応したときの失敗を恐れ、多くの場合が事後的な対応になっているのが 現状であると考える。開発スピードを優先して考えれば、どちらの対応が好ましい かは、すでに答えの出ているところであると考える。 33 3.5 国内開発における Translational Research (医師の自主臨 床研究) の活用 我が国では、医師の自主臨床研究により、国内外未承認薬の臨床研究が可能となる 場合がある。もちろん、薬事法規制下のいわゆる“治験”とはその性格は異なるた め、本臨床研究のデータを製造販売承認申請時の評価資料として用いることはでき ない。また、薬事法との関係から、国内企業で製造された未承認薬を開発企業が医 師に提供し、その医師が自主臨床研究は行うことはできない。医師の自主臨床研究 に用いることができる未承認薬は、医師の個人輸入品又は院内製造品のみである。 このような条件下ではあるが、医師の自主臨床研究が創薬系バイオベンチャーの開 発意思決定よりも先行する場合は、この臨床研究のデータは非常に貴重なヒトでの データとなる。この臨床研究の成績と品質次第では、薬事法規制下の治験で同じ試 験を繰り返すことなく、次の段階の臨床試験に移行することが可能となる場合もあ る。事実、筆者は、特殊な開発領域ではあるが、医師の自主臨床研究のデータを活 用し、国内第3相臨床試験(治験)及び海外第2相臨床試験(治験)への移行に成 功している。すなわち、我が国の医師の自主臨床研究の仕上げ方によっては、この ような開発も可能なのである。 更に筆者は、医師の自主臨床研究の成果を活用し、次相の治験に移行するという開 発戦略が特殊領域の特殊例で終わらないために、規制当局主催により半期に一度開 催される「新薬審査部門定期説明会」で規制当局に文書で質問を行い、その質問に 対し、厚生労働省医薬食品局審査管理課が医薬品研究という国内専門誌に文書で回 答を示してくれた。図 3 にその内容の概略を示す。 34 図 3 医師の自主臨床研究の成果の活用及び実施対応について 詳細は、 「医薬品研究 Vol.34, No.9, 2003」をご参照いただきたい 8)。このとき に既に Translational Research の成果の社会還元の道筋が示されている。また、 医師の自主臨床研究の成果が、海外治験の移行にも役立つ場合もあるため、その臨 床研究の品質をしっかりと確保することを前提に研究を行えるようなインフラ構 築が更に進むことを期待している。 一方で、医師の自主臨床研究については、治験を管轄する規制当局が試験登録する 実施形態ではないため、この研究体制自体への批判も存在する。医薬品開発の王道 は、法規制に従った治験実施であることは間違いないことから、医師の自主臨床研 究そのものをベンチャー企業の事業計画の主軸に置く開発戦略は避けたい。医師の 自主臨床研究は、研究そのものが目的であり、当該医薬品の製造販売承認を獲得す ることが目的ではない。当該医薬品が社会還元される際の有意義な参考データと考 えるべきである。新薬開発における医師の自主臨床研究の位置づけについては、未 だその社会的なコンセンサスや法的な位置づけも明確化されている状況ではない と考える。我が国における医師の自主臨床研究を推進については、賛否両論が存在 することからも、当該臨床研究の成果を事業に活用する際には、慎重に考えていき たい。また、医師の自主臨床研究の成果を事業に活用するとしても、臨床研究開始 35 前に対応すべきことも少なくないため、Translational Research の推進に力を入 れている大学等での臨床研究実施が望まれるところである。 ここで強調しておきたいことは、医師の自主臨床研究が創薬系バイオベンチャーの 開発の主軸になることはないということである。もし、創薬系バイオベンチャー企 業の開発品目による臨床研究が先行していたとしても、当該ベンチャー企業はしっ かりと治験準備を進めることを前提に開発を進めるべきであると考える。 36 3.6 海外での早期 POC 臨床試験 これまで繰り返し述べてきたが、我が国の創薬系バイオベンチャーが開発戦略を構 築する際は、事業計画にも十分に配慮する必要がある。資金の限られているベンチ ャー企業とって、事業計画が企業運営の根幹となり、開発戦略はその事業戦略を具 現化する最も大きなものとなる。事業戦略プラス費用対効果を勘案しながら開発戦 略を構築し、早期 POC 臨床試験を実施する計画を立案する必要がある。これらに 忠実に対応しようとすると、これまでにも述べてきたように海外での POC 臨床試 験の実施を優先的に考えることになる。企業理念として、国内シーズの国内優先還 元等をポリシーとして、企業運営するようなベンチャーは別として、ほとんどが海 外開発先行型になると思われる。この流れは、ビジネス理論としては、当然なのだ ろう。 海外での臨床試験の際に、大きな問題となるのが、いわゆる“丸投げ型”の試験委 託である。この“丸投げ型”に再三警笛を鳴らしているが、以下にその主な理由を 示す。 POC 臨床試験の多くはオープンラベル試験で行われ、そのデータをリアル タイムで収集することも可能である。この試験段階では、被験薬のあらゆる ポテンシャルを探るために、最善を尽くさなければならない。臨床試験実施 中に、その試験データを確認しながら、用量変更したり、投与レジメン変更 したり、試験方法を変更したりすることもあるかもしれない。 POC 臨床試験は非常にデリケートな試験であることから、被験薬の特性等 をしっかりと把握している開発担当者が、常に緊張感を持って対応すること が望まれる。したがって、POC 臨床試験において“丸投げ型”は好ましい と言うことはできず、CRO に委託したとしても共同実施の意識を高く持っ て対応することが必要であると考える。 事実、第3回の「ヒト POC に関する勉強会」で招聘したアステラス製薬臨 床管理部の津田課長からは、アステラス製薬では POC 臨床試験は CRO 委 託せずに自社対応しているとのお話であった。大手製薬企業においても、デ リケートな POC 臨床試験を CRO に外注することは、現時点では考えてい ないとのことであった。第 3 回の勉強会では、大企業から学ぶところも多 37 いことを実感した。 我が国の創薬系バイオベンチャーによる POC 臨床試験は、その多くが海外 で行われることになるであろう。予定どおりに開発を進めるためにも、当該 試験を依頼する CRO には予定どおり試験を完結してもらわなければなら ない。CRO における当該ベンチャー企業の臨床試験の優先順位を向上させ る努力を怠ってはならない。CRO にとって、いわゆる“良いお客さん”と いわれるような存在にならないようにする。 上述のような意識を持っていると、いわゆる“丸投げ型”の臨床試験の依頼になる ことはないと思われる。POC 臨床試験は、 “手がかかる(手をかける)”ことを意 識した対応を心掛けたい。 38 3.7 日米欧における臨床試験の開始 日米欧各極(各国)により、治験申請時の相談方法、要求事項、治験申請後の審査 期間及び実施医療機関での審査体系等が異なる。開発領域により、first in human trial までに、より厳しい審査を求められるバイオ医薬品も存在する(遺伝子治療用 医薬品、細胞・組織利用医薬品等)。本手引書で、開発領域ごと、各国ごとの治験 実施までの流れをそれぞれ述べることは、莫大な情報量になることから記載を控え るが、日米 EU での地域に分け、その概略を述べさせていただきたい。 「ヒト POC に関する勉強会」の参加者が多数興味を示した米国の治験開始までの流れについて は、多少詳しく述べさせていただきたい。 米国での治験 3.7.1 1) IND (Investigational New Drug) 申請 米 国 に お け る 治 験 申 請 は 、 IND 申 請 ( Investigational New Drug Application)と呼ばれ、米国食品医薬品局(U.S. FDA, Food and Drug Administration)の所定の様式を用い、当該医薬品の審査担当部門宛てに IND 申請しなければならない。IND カテゴリーは、以下の 2 カテゴリーがある。 Commercial Research (Non-commercial) 通常、企業がスポンサーとなる治験申請は、Commercial IND と呼ばれる。 また、Commercial IND の他、IND のタイプもいくつか存在し、そのタイプ を以下に示す。 2) Investigator IND Emergency Use IND Treatment IND IND 申請資料 通常、以下の関連資料を含め、IND 申請資料を作成する必要がある。 39 3) Animal Pharmacology and Toxicology Studies Manufacturing Information (CMC information) Clinical Protocols and Investigator Information IND 審査期間 FDA の審査担当部門による 30 days review (calendar days)が行われる。 当該 IND に対し疑義照会がない場合は、この期間経過後に治験を開始するこ とができる。疑義照会がある場合でも、この 30 日以内に解決されれば、この 期間後に治験開始が許可される。この 30 日間で疑義照会が終了しない場合又 は追加試験等を求められた場合は、 “Clinical Hold”を宣言する FAX(書簡) が申請者に届き、この hold issue が解決されるまで、治験開始は認められな い。申請者が“Clinical Hold”の hold issue に対する回答書を当該 FDA 審 査担当部署に提出後は、再度、30 days review の対象となる。 4) Pre-IND Meeting IND 申 請 す る 前 に 、 申 請 者 は 当 該 FDA 審 査 担 当 部 署 に よ る Pre-IND Consultation を受けることができる。多くの場合は電話会議での対応のよう であるが、face-to-face での会議を行う場合もある。申請者から書面で Pre-IND Meeting の開催要請がされてから、 60 日以内に会議が開催される。 また、Pre-IND Meeting の information package(3.7.1 項 5)で後述)は、 予定されている会議日の少なくとも4週間前までに提出することになってい る。 FDA による Pre-IND Meeting は、first in human trial に向けた早期の問題 抽出及び問題解決のための位置づけにあり、Pre-IND Meeting での課題をし っかりと解決し、IND 申請を行うことが 30 days review をスムースに乗り 越えるためには大切である。しかし、著者は Pre-IND Meeting で問題提起さ れなかったことが、IND 申請後の 30 days review で提示された経験を何度 か持っている。特に、不安要素を含む案件については(特に、考え方が画一的 でない案件) 、Pre-IND Meeting でしっかりと確認する方向で対応することが 良いであろう。 なお、Pre-IND Meeting の正式な議事録は、会議後 30 日以内に申請者に提 供される。 40 5) Information Package Pre-IND meeting の“Information Package”として、一般的に必要とされ る情報を以下に示す。 6) 製品名及び申請番号 化学名及び構造 予定される適応症 剤型、投与経路及び投与方法(投与頻度及び期間) 会議目的の概略陳述(以下の内容を含む) − 終了した試験 − 予定されている試験 − 会議で協議するデータ − 重要な相談事項の本質 − 全体的な開発計画 主な会議目的のリスト及び会議から期待される結果 会議の議事(案)… 各議事に必要な協議時間及び説明者 分野ごとにグループ化された特別な質問事項リスト 臨床試験データの概略 非臨床試験データの概略 CMC information の概略 Pre-IND Meeting 及び IND 申請の費用 現時点では、Pre-IND Meeting 及び IND 申請に関わる費用は無料である。 7) IND 申請に関するガイダンス 以下に IND 申請に役立つガイダンスを示す。 Guidance for Industry: INDs - Approaches to Complying with cGMP's for Phase 1 Drugs (Draft) (Jan. 2006) Guidance for Industry: Exploratory IND Studies (Jan. 2006) Content and Format of Investigational New Drug Applications (INDs) for Phase 1 Studies of Drugs Including Well Characterized, Therapeutic, Biotechnology-Derived Products (Nov. 1995) Q & A - Content and Format of INDs for Phase 1 Studies of Drugs, Including 41 Well-Characterized, Therapeutic, Biotechnology-Derived Products (Oct. 2000). Bioavailability and Bioequivalence Studies for Orally Administered Drug Products - General Considerations (July 2002). IND Exemptions for Studies of Lawfully Marketed Drug or Biological Products for the Treatment of Cancer (Jan. 2004) Drug Master Files (Sept. 1989) Required Specifications for FDA's IND, NDA, and ANDA Drug Master File Binders (Apr. 1998) 8) Immunotoxicology Evaluation of Investigational New Drugs (Oct. 2002). Institutional Review Board(IRB)及び Institutional Biosafety Committee (IBC) 我が国と同様に、米国でも実施医療機関に設置される IRB(Institutional Review Board; 施設内治験審査委員会)での審査・承認を経て、治験担当医 師との契約を締結し(日本では実施医療機関との契約)、被験者への被験薬投 与が可能となる。IRB の開催タイミングは、1個月に1回の施設がほとんどの よ う で あ る 。 ま た 、 recombinant DNA research を 行 う 際 に は 、 NIH (National Institute of Health; 米国国立衛生研究所)の定めるガイドライ ンに従い、その研究機関に IBC (Institutional Biosafety Committee; 施設 内生物学的安全性委員会)を設置し、審査することが求められている。したが って、米国で recombinant DNA research に相当する治験を行う場合は、 実施医療機関の IRB と IBC の2つの委員会の実施承認を得る必要がある。筆 者の経験からは、IBC のほうが IRB よりも専門家による審査や関連規制への適 合確認等がより慎重に行われているように感じられ、より厳格な審査になって いるように思われる。 42 3.7.2 欧州での治験 欧州では、2004 年に発効された欧州臨床試験指令(EU Clinical Trial Directive, Directive 2001/20/EC)により、EU 加盟国では、基本的に 1 加盟国で 1 つの Ethics Committee (EC)での承認及び当該国の規制当局の治験実施許可があれば 臨床試験の実施が可能となった。EC と規制当局は、基本的に治験申請後 60 日以 内に治験実施の可否判断を行わなければならず(遺伝子治療、細胞治療、遺伝子組 換え医薬品は 90 日以内、異種移植は無制限) 、EC からの照会事項は申請後、35 日以内に申請者に出され、その解答は EC で再確認される。もし、この再確認の結 果、実施不可の通知が出された場合は、再度、一から申請をやり直すことになる。 また、規制当局審査と EC 審査は、一般的に並行して対応することが可能である。 したがって、我が国や米国のように、治験実施にあたり、各実施医療機関での IRB 審査がなく(EC による中央審査体験が確立している)、非常に効率的な対応とな る。 一方で、EU Clinical Trial Directive は存在するが、各国で独自の治験申請や EC 申請の審査期間の期限を設定していることから、EU 加盟国の詳細な状況について は、それぞれ確認しなければならない。また、臨床試験の相や単施設試験/多施設 試験の違いで審査期間が異なったり、条件により審査期間が短縮したりする場合も ある。Competent Authority 及び EC の審査料は各国により異なる。 参考までに、EU 諸国の治験申請審査に関わる情報を表 2 にまとめた。 43 表 2 Country EU 諸国の治験申請審査に関わる状況 Competent Authority Ethics Committee AUSTRIA 35 days 35 days BERGIUM Ph.1: 15 days Ph.1: 15 days Ph.2-4: 28 days Ph.2-4: 28 days CZECH REPUBLIC 60 days 60 days DENMARK 30 working days/6 weeks 60 days FINLAND 60 days 60 days max. FRANCE 60 days 60 days GERMANY 30 day GREECE 60 days 60 days HUNGARY 60 days 60 days IRELAND 30 days 60 days ITARY Ph. 1: 30-90 days 60 days 60 days (Multicenter trial) 30 days (Single center trial) Ph. 2-3: 60 days Ph. 4: 60 days LUXEMBURG 60 days max. 60 days max. THE NETHERLANDS Ph. 1: 21 days max. 60 days Ph. 2-4: 49 days max. PORTUGAL 60 days 60 days SLOVAKIA 60 days max. 60 days max. SPAIN 60 days 60 days SWEEDEN 60 days or 30 days 60 days max. (aiming dependent) GMP certificate from 3 rd countries UNITED KINGDOM Ph. 1: 14-21 days Ph. 2-4: 30 days 44 60 days 3.7.3 日本での治験 基本的に、米国の治験申請審査システムに近い。遺伝子治療用医薬品や細胞組織利 用医薬品等の特殊なケースを除き、治験申請後、PMDA(Pharmaceuticals and Medical Devices Agency; 独 立 行 政 法 人 医 薬 品 医 療 機 器 総 合 機 構 ) に よ る 30days review を経てからでないと、実施医療機関との治験契約はできない。こ の 30days review で審査が終了しない場合は、米国のような”Clinical Hold”は宣 言されることなく、規制当局と申請者による照会事項回答が繰り返される。実施医 療機関には、各々IRB が設置され、それぞれ独自の審査を行う(中央 IRB 制度も検 討されている) 。治験申請と IRB 申請を同時並行で行うこともできるが、治験申請 通過後、実施医療機関の IRB 申請を行っているのが実情のようである。 医 薬 品 第 1 相 試 験 開 始 前 相 談 ( 米 国 の Pre-IND Meeting に 相 当 ) に は 、 \4,329,400 の費用がかかる。治験申請費用は無料である。 45 3.7.4 アジア諸国での POC 臨床試験の可能性 創薬系バイオベンチャーが製薬企業とのアライアンス確度を向上させるためにも、 欧米での POC 臨床試験が第一選択として考慮されるであろうことをこれまで述べ てきたが、アジア諸国に患者が多い肝疾患(HBV, HCV, 肝がん等)、HIV 及び急 性感染症等については、そのマーケットポテンシャルからアジアでの POC 臨床試 験の可能性が十分にある。事実、米国のバイオベンチャーによる肝疾患領域でのア ジアでの POC 臨床試験のニーズは存在する。また、将来的なアジア諸国の経済発 展を考えると、人口の約過半数を占めるアジアの潜在的なマーケットは魅力のある ものになることから、コストベネフィットがあり、スピード感があるアジアでの POC 臨床試験はこれから注目されることになるであろう。 しかし、現時点で、アジア諸国で実施されている臨床試験は、その多くが後期臨床 試験の国際共同治験となり、POC 臨床試験数は非常に少ない。探索的試験に位置 づけられる POC 臨床試験は、安全性プロファイルがまだ十分に把握された状況に あるとは言い難く、特に、バイオ医薬品の場合、リスクベネフィット勘案のうえで 試験が成立していることもあり、臨床試験のマネジメントという観点からは、CRO に“丸投げ”するような性格の試験ではない。多くの場合、被験薬のプロファイル を熟知している企業の Hands-on フォローアップが必要となる。特に、急性増悪 する疾患やがん等の試験は、慎重対応が要求される。アジア諸国での POC 臨床試 験は、これからその実施体制が本格的に整備されると思われる。 また、遺伝子組換え医薬品やバイオ医薬品に関わる法規制の整備が途上段階にある 国も多く、十分に法規制を確認することも忘れてはならない。治験審査のスピード は、早いと思われがちだが、それまでの審査経験値に依存する部分もあるため、一 概に“早い”と決めつけるのは危険である。当該国の薬事規制をよく熟知している コンサルタントの支援は必要になるであろう。 46 4. まとめ 我が国の創薬系バイオベンチャーにおける早期 POC 確保のための留意点を述べて きたが、最も大切なことは、事業開発戦略と研究開発戦略のバランスを常に意識す ることである。どちらかが変更を余儀なくされた場合は、すぐに双方のバランスを 取り直すことが必要である。資金調達計画と研究開発の成果のマイルストンは、密 接な関係にあることからも、このことは自明である。 また、我が国の創薬系ベンチャー企業に見落とされがちなのが、開発薬事の重要性 である。事業開発や開発戦略を構築する際に、創薬開発の原理原則となる法規制や ガイダンス等の理解なくして現実的な戦略は策定できない。開発経験の少ない未知 な分野であればあるほど、開発薬事は重要である。開発領域によっては、規制自体 が成長段階にある場合もあり、リアルタイムにその変化に対応していくことが求め られる。また、国内や米国の法規制やガイダンス等は、比較的分かりやすいが、欧 州の薬事規制は、EU 諸国では EU Clinical Trial Directive をもとに各国独自の規 制が存在し、EU 加盟国以外の欧州諸国も存在することから、非常に複雑である。 海外での臨床試験を選択する際は、特にバイオ医薬品の場合、当該開発国の開発薬 事の専門家の支援は必須であろう。開発薬事の専門家は、臨床試験実施国の規制当 局と密接したコミュニケーションを取ることができるコンサルタント等が望まし い。また、開発者自身も実施国の薬事事情は、ある程度把握しておく必要があると 思われる。海外では、規制当局の審査官が当局退職後、コンサルタントとして活躍 しているケースも散見されるため、このような辣腕コンサルタントを選択したい。 大手海外 CRO には、このような薬事コンサルタントが存在するが、中小規模の海 外 CRO の場合には事前に薬事対応の専門家のバックグラウンドや存在自体をしっ かりと確認しておく必要がある。 さらに、 “丸投げ型”の海外 POC 臨床試験の実施は好ましくないことから、自社 でしっかりと臨床試験をコントロールすることも忘れてはいけない。自社に開発の 専門家がいない場合は、国内の開発アドバイザーによる支援も積極的に検討したほ うがよいであろう。この場合は、できる限り、薬事に強い臨床開発の専門家が良い。 POC 臨床試験の実施に向け、自社の強みと弱みをしっかりと分析し、弱みに対し ては早期に能力の高い外部支援を検討することが、早期 POC 試験の実施を可能と し、長い目で見た場合は、コストベネフィットにも結び付くのであろう。もちろん 47 質の良い外部協力者を見抜く力は必要となる。 また、画一的な考え方や“あるべき論”に縛られず、常に柔軟性に富んだケースバ イケースの対応を心掛け、臨床試験実施規国の規制当局と密にコミュニケーション のとれる環境を作ることが大切である。 POC 臨床試験の中心は当該開発企業に置き、その企業の“弱み”は積極性が高く 信頼できる外部支援で補い、開発を進めることが理想的であろう。我が国の創薬系 バイオベンチャーの創業には、シーズの発明者となる医師や研究者が参加している ケースも多いことから、国内のアカデミアのパワーを積極的に使うことも戦略の一 つとして忘れてはならない。 産学連携で誕生したベンチャーが、“官”に近づいたときに、真の産官学連携がス タートする。この“官”は、 “産”と“学”の間にあることが大切である。POC 臨 床試験の場合は、しばらくは海外の“官”が多くなるのかもしれない。社内リソー スの少ないベンチャー企業にとって、規制当局を含む強い外部ネットワーク形成は、 早期 POC 試験を実現するばかりでなく、長期的な企業発展のためにも大切なこと であると考える。 我が国の創薬系バイオベンチャーにおいて早期 POC 確保するための留意点を中心 に述べてきたが、この留意点も時代とともに変化していく可能性がある。普遍的な 原理原則を主軸に、変化には柔軟に対応し、スピード感を最も大切に創薬開発に臨 むマインドを持ち続けていきたい。 48 参考文献等 1. 「2006 年バイオベンチャー統計調査報告書」 (財団法人バイオインダストリー協会/編), 財 団法人バイオインダストリー協会, 2007 2. 小澤健夫, バイオテクノロジージャーナル, 7(1), 108-111, 2007 3. ICH Q5A, ヒト又は動物細胞株を用いて製造されるバイオテクノロジー応用医薬品のウ イルス安全性評価, 厚生省医薬安全局審査管理課長通知, 医薬審第 329 号, 2000 年 2 月 22 日 ICH Q5B, 組換え DNA 技術を応用したタンパク質生産に用いる細胞中の遺伝子発現構 成体の分析, 厚生省医薬安全局審査管理課長通知, 医薬審第 3 号, 1998 年 1 月 6 日 ICH Q5C, 生物薬品(バイオテクノロジー応用製品/生物起源由来製品)の安定性試験, 厚 生省医薬安全局審査管理課長通知, 医薬審第 6 号, 1998 年 1 月 6 日 ICH Q5D, 生物薬品( バイオテクノロジー応用医薬品/生物起源由来医薬品) 製造用細 胞基材の由来、調製及び特性解析, 厚生省医薬安全局審査管理課長通知, 医薬審第 873 号, 2000 年 7 月 14 日 ICH Q5E, 生物薬品(バイオテクノロジー応用医薬品/生物起源由来医薬品)の製造工程 の変更にともなう同等性/同質性評価, 厚生労働省医薬食品局審査管理課長通知, 薬食審 査発第 0426001 号, 2005 年 4 月 26 日 4. ICH Q6B, 生物薬品(バイオテクノロジー応用医薬品/生物起源由来医薬品)の規格及び試験 方法の設定, 厚生労働省医薬局審査管理課長通知, 医薬審発第 571 号, 2001 年 5 月 1 日 5. ICH M3, 医薬品の臨床試験のための非臨床安全性試験の実施時期についてのガイドライン, 厚生省医薬安全局審査管理課長通知, 医薬審第 1019 号, 2000 年 11 月 13 日(改正) 6. ICH S6, バイオテクノロジー応用医薬品の非臨床における安全性評価, 厚生省医薬安全局審 査管理課長通知, 医薬審第 326 号, 2000 年 2 月 22 日 7. 厚生労働省, 遺伝子治療用医薬品の品質及び安全性の確保に関する指針, 厚生労働省医薬 食品局長通知, 薬食発第 1228004 号, 2004 年 12 月 28 日(一部改正) 厚生労働省, ヒト由来細胞・組織加工医薬品等の品質及び安全性の確保に関する指針, 厚 生労働省医薬安全局長通知, 医薬発第 1314 号, 2000 年 12 月 26 日 8. 林亜紀子, 医薬品研究, 34(9), 591-601, 2003 49 首都圏バイオネットワークとは 国内バイオ産業の発展のために、経済産業省主導のもと形成した、首都圏を中心と するバイオベンチャー支援のプラットフォームです。 バイオベンチャーをはじめ、大手企業や大学・研究機関など、多くのバイオ関連企 業・機関が参画し、ネットワークを形成しています。 大手企業とのアライアンス促進、知財戦略・資金調達など多様なテーマを設定した 研究会・セミナーの開催、専門家の派遣など、バイオベンチャーの抱える課題に応 じて様々な支援を行っています。 詳細は下記ホームページをご覧ください。 http://www.shutoken-bio.net/ 事 務 局 財団法人バイオインダストリー協会 104-0032 東京都中央区八丁堀2-26-9 グランデビルディング TEL. 03-5541-2731 FAX. 03-5541-2737 URL http://www.jba.or.jp/ 経済産業省 関東経済産業局 地域経済部地域経済課 330-9715 バイオ産業チーム 埼玉県さいたま市中央区新都心1-1 さいたま新都心合同庁舎 1 号館 10F TEL. 048-600-0342 FAX. 048-601-1293 URL http://www.kanto.meti.go.jp/ 8F - 非売品 - 禁無断転載 創薬開発において 早期 POC 確保するための手引き 発 行 発行者 平成20年3月 財団法人バイオインダストリー協会 東京都中央区八丁堀2-26-9 グランデビルディング 電話 03-5541-2731 8F
© Copyright 2026 Paperzz