Title
Author(s)
配位環境を制御した単核銅-酸素活性種の合成とその反応
性
藤井, 達也
Citation
Issue Date
URL
2007
http://repo.lib.nitech.ac.jp/handle/123456789/131
Rights
Type
Textversion
Thesis or Dissertation
author
・名古屋工業大学学術機関リポジトリは、名古屋工業大学内で生産された学術情報を
電子的に収集・保存・発信するシステムです。
・論文の著作権は、著者または出版社が保持しています。著作権法で定める権利制限
規定を超える利用については、著作権者に許諾を得てください。
・Textversion に「Author」と記載された論文は、著者原稿となります。
実際の出版社版とは、レイアウト、字句校正レベルの異同がある場合もあります。
・Nagoya Institute of Technology Repository Sytem is built to collect, archive and
offer electronically the academic information produced by Nagoya Institute of
Technology.
・The copyright and related rights of the article are held by authors or publishers.
The copyright owners' consents must be required to use it over the curtailment of
copyrights.
・Textversion "Author " means the article is author's version.
Author version may have some difference in layouts and wordings form publisher
version.
博士論文
配位環境を制御した単核銅-酸素活性種の
合成とその反応性
2007 年
藤 井
達 也
目次
第1章
緒言
はじめに
1
1.1
酸素分子とその活性化
1
1.2
銅を含有する機能性タンパク質・酵素
2
1.3
DβM, PHM
3
1.4
錯体による銅-酸素会合体の合成
5
1.5
研究の目的
8
References
第2章
10
酵素の反応機構に基づいた単核銅(II)-ハイドロパーオキソ錯体の合成
2.1
序論
17
2.2
実験
18
2.2.1
配位子合成
18
2.2.2
錯体合成
18
2.2.3
単核銅(I)錯体と酸素分子との反応
18
2.2.4
反応基質
18
2.2.5
測定装置
19
2.3
21
結果
I
+
21
I
+
2.3.2 [Cu (H2bppa)] 錯体と酸素分子との反応
21
2.3.3
銅(II)錯体と過酸化水素の反応系との比較:反応機構の検討
22
2.3.4
スピントラップ剤を用いた単核銅(II)-スーパーオキソ錯体の捕捉
23
2.3.1
2.4
[Cu (H2bppa)] 錯体の構造
25
考察
2.4.1
単核銅(II)-スーパーオキソ錯体の合成と観測の試み
2.4.2
単核銅(II)-スーパーオキソ錯体を経由した
単核銅(II)-ハイドロパーオキソ錯体の合成
2.5
25
26
結論
28
References
29
第3章
単核銅(II)-スーパーオキソ錯体による水素原子引き抜き能の検討
3.1
序論
39
3.2
実験
40
3.2.1
3.2.2
40
配位子合成
I
+
[Cu (bnpa)] 錯体の合成
40
I
3.2.3
単核銅(I)錯田と酸素分子との反応
40
3.2.4
外部基質
40
3.2.5
測定機器
41
3.3
44
結果
I
+
3.3.1
[Cu (bnpa)] 錯体の合成と酸素分子との反応
44
3.3.2
スピントラップ剤による反応中間体の検出
45
3.3.3
基質存在下における銅(I)錯体と酸素分子との反応
45
3.3.4
単核銅(II)-スーパーオキソ種の水素原子引き抜き能の検討
47
3.4
48
考察
I
+
3.4.1
[Cu (bnpa)] 錯体と酸素分子との反応生成物
48
3.4.2
分子内アミド基が酸素との反応性に与える影響
49
3.4.3
単核銅(II)-スーパーオキソ錯体の反応性
49
3.5
結論
51
References
52
第4章
平面 4 配位型の単核銅(II)-ハイドロパーオキソ錯体の合成と反応性の検討
- 5 配位型錯体との基本的物性ならびに反応性の比較 -
4.1
序論
61
4.2
実験
62
4.2.1
配位子合成
62
4.2.2
錯体合成
62
4.2.3
銅(II)錯体と過酸化水素との反応
64
4.2.4
酸化反応実験
64
4.2.5
触媒反応
64
4.2.6
測定装置
64
4.3
68
結果
4.3.1
4.3.2
4.3.3
4.3.4
II
2+
II
2+
II
2+
[Cu (bpba)(MeOH)] , [Cu (bpba)(MeCN)2] , [Cu (bpba)(H2O)] 錯体の構造
II
2+
[Cu (bpba)(X)] 錯体溶液の分光学的測定 (X=MeOH, MeCN, H2O)
68
68
II
2+
-
69
II
2+
-
69
[Cu (bpba)(MeOH)] 錯体と N3 イオンとの反応
[Cu (bpba)(MeOH)] 錯体と Cl イオンとの反応
II
2+
4.3.5
[Cu (bpba)(H2O)] 錯体と過酸化水素との反応
70
4.3.6
単核銅(II)-ハイドロパーオキソ錯体の熱力学的安定性
71
4.3.7
単核銅(II)-ハイドロパーオキソ錯体と基質との反応
71
4.3.8
触媒的酸化反応
72
4.4
74
考察
4.4.1 4 配位型単核銅(II)-ハイドロパーオキソ錯体の合成
II
74
4.4.2
4.4.3
4.5
[CuII(bpba)(OOH)]+錯体の基本的物性
II
+
[Cu (bpba)(OOH)] 錯体の反応性
74
75
結論
77
References
78
第5章
異なった平面性を有する単核銅(II)-ハイドロパーオキソ錯体の合成
- 4 配位型錯体間の構造と反応性の相関 -
5.1
序論
93
5.2
実験
94
5.2.1
配位子合成
94
5.2.2
錯体合成
95
5.2.3
銅(II)錯体と過酸化水素との反応
97
5.2.4
触媒反応
97
5.2.5
測定装置
97
5.3
101
結果
5.3.1
5.3.2
5.3.3
5.3.4
5.3.5
単核銅(II)錯体の構造と分光学的性質
2+
-
102
II
2+
-
103
[Cu (Lsq)(MeOH)] 錯体と N3 イオンとの反応 ; (Lsq =bpba, bpipa, bpea)
[Cu (Lsq)(MeOH)] 錯体と Cl イオンとの反応 ; (Lsq =bpba, bpipa, bpea)
II
2+
[Cu (Lsq)(X)] と過酸化水素との反応 ; (Lsq = bpba, bpipa, bpea)
104
II
2+
105
II
2+
105
[Cu (Lsq)(OOH)] 錯体の熱力学的安定性 ; (Lsq =bpba, bpipa, bpea)
5.3.6 [Cu (Lsq)(OOH)] 錯体による触媒反応
5.4
101
II
107
考察
5.4.1
アルキルアミン置換基と錯体の基本的物性の関係
107
5.4.2
各銅(II)錯体と過酸化水素との反応
108
5.4.3
各錯体の反応性の比較
109
5.5
結論
110
References
111
第6章
4 座型配位子を用いた単核鉄(III)-ハイドロパーオキソ錯体の
合成とその触媒反応性の検討
6.1
序論
125
6.2
実験
127
6.2.1
配位子合成
127
6.2.2
錯体合成
127
6.2.3
鉄(II)錯体と過酸化水素との反応
128
6.2.4
触媒反応
128
III
6.2.5
6.3
128
測定装置
131
結果
6.3.1
II
131
II
Fe -PPBA 錯体の合成と構造
6.3.2
Fe -PPBA 錯体と過酸化水素との反応
131
6.3.3
過酸化水素を酸化剤とした触媒反応
132
6.4
6.4.1
6.4.2
6.5
133
考察
単核鉄(III)-ハイドロパーオキソ錯体の合成と性質
III
2+
[Fe (ppba)(OOH)] 錯体の酸化反応特性
133
133
結論
135
References
136
第7章
143
総括
発表論文
145
謝辞
147
IV
訳語
DβM : Dopamine β-monooxygenase
PHM : Peptidylglycine α-hydroxylating monooxygenase
SOD : Superoxide dismutase
PAL : peptidylamidoglycolate lyase
TPA : tris(2-pyridylmethyl)amine
TREN : tris(2-aminoethyl)amine
TMG3-TREN : tris(tetramethylguanidino)tren
HB(3,5-Me2(pz))3 : hydrotris(3,5-dimethyl-1-pyrazolyl)borate
HB(3-tBu-5-iPrpz)3 : hydrotris-(3-tert-butyl-5-iso-propylpyrazolyl)borate
HB(3,5-iPr2pz)3 : hydrotris(3,5-di-iso-propyl-1-pyrazolyl)borate
XYL-O- : 2,6-bis[bis[2-(2-pyridyl)ethyl]amino]phenolate
Py2SSPy2 : bis{2-[N,N-bis(3-pyridylmethyl)-amino]-1,1-dimethylethyl}disulfide
Hbdpi : 4,5-bis[di(2-pyridylmethyl)aminomethyl]imidazole
H2BPPA : bis(6-pivalamido-2-pyridylmethy)(2-pyridylmethyl)amine
BNPA : bis[(6-neopentylamino-2-pyridyl)methyl][(2-pyridyl)methyl]amine
BPBA : N,N-bis (2-pyridylmethyl)-tert-butylamine
BPIPA : N,N’-Bis(2-pyridylmethyl)iso-propylamine
BPEA : N,N’-Bis(2-pyridylmethyl)-ethylamine
N3S : 2-bis(6-methyl-2-pyridylmethylethyl)amino-1-(phenylthio)ethane
MPPA : bis(2-pyridylmethyl) (6-pivalamido-2-pyridylmethy)amine
PPBA : (6-pivalamide-2-pyridylmethyl)(2-pyridylmethyl)tert-butylamin
DMPO : 5,5-dimethyl-1-pyrroline-N-oxide
TEMPO-H : 2,2,6,6-tetramethyl-1-hydroxypiperidine
Guaiacol : 4-methoxyphenol
BDE : bond dissociation energy
r.d.s. : rate determining step
ET : electron-transfer,
PT : proton-transfer
HAT : hydrogen atom transfer
V
第1章
緒言
はじめに
地球上に存在する好気性生物は呼吸により酸素分子を体内に取り込み、摂取した栄養素
を段階的な酸化反応により分解させることでエネルギーを取り出し、生命を維持している1。
生体内に取り込まれた酸素分子の役割の一つは、そのエネルギー獲得プロセスにおける電
子受容体としての働きであり、最終的に酸素分子は四電子還元され水となる。一方、取り
込まれた酸素分子は、各種基質に対する直接的な酸素原子の挿入反応に用いられることも
見いだされている。このような反応により、体内では生命の維持に必要な物質変換を行っ
ている2。これらの反応は体内に存在する酵素により触媒され、制御されている。酵素は巨
大なタンパク質であり主に C, H, N, O などの有機元素から構成されているが、酸素添加酵素
の多くはその活性中心に金属イオンを含んでいる。金属イオンは幅広い酸化数をとること
ができるため電子移動の中心として、また配位結合による小分子の捕捉・活性化の中心と
して機能しており3、酸素分子の活性化には必要不可欠である。酵素はアミノ酸残基による
配位結合により活性中心に金属イオンを捕捉しており、先に述べた金属特有の性質と巨大
分子である酵素の空間的制御により、高い反応性や基質特異性を発現している 3。酵素の金
属中心における酸素分子の活性機構を知ることは生化学的な立場から非常に有用であり、
得られた知見は新たな機能性分子の創成に大きく貢献すると考えられる。
1.1
酸素分子とその活性化
基底状態の酸素分子は 2 つの縮重した反結合性π*軌道(π*x, π*y)にそれぞれ不対電子を持
った常磁性の等核二原子分子である。これらの不対電子はスピン平行にあることから基底
状態の酸素分子は三重項(3Σg-)であり、通常の一重項分子との反応はスピン禁制であるため、
その反応性は大きく抑制される4。しかしながら、炭化水素と酸素分子との反応は熱力学的
には大きな発熱反応であり、最終的に炭化水素は二酸化炭素まで酸化され、酸素は水まで
還元された状態がエネルギー的に最も安定である。すなわち、酸素分子は通常の有機物と
はスピン禁制であるため直接反応することができないが、反応が進行すれば大きなエネル
ギーが得られるという性質を有している。酸素分子が関与するスピン許容な反応は以下の 3
つに分類できる。
1
(i) 一重項酸素(1∆g)との反応
三重項酸素が励起され 1 つの不対電子がスピン反転した状態。エネルギーの高い 1Σg+を経
由したのち 1∆g となる。一重項であるため一般的な有機物との反応はスピン許容であり、片
方のπ*軌道が完全に空であるため求電子的な付加反応が進行する。
(ii) ラジカル反応
ラジカル分子は二重項であるため、三重項酸素との反応で二重項分子を生成する反応は
スピン許容である。酸素分子はビラジカルであるため、このような反応は拡散律速に近い
速度で進行する。アルキルラジカルによる自動酸化反応や物質の燃焼反応がこれにあたる。
(iii) 電子移動反応
一重項電子供与体(D)から三重項酸素への電子移動反応は、三重項ラジカルイオンペアを
生成するためスピン許容である。このような過程を経て酸素分子が還元される。
1
D + 3O2 → 3(D・+O2・-) → D・+ + O2・-
(i)の反応には光励起が必要であり、(ii)の反応にはエネルギーの高いラジカル種の生成が
必要であるため、(iii)が我々の生体内における酸素分子の活性化様式である。一般的に酸素
分子の還元種はそれ自身よりも酸化還元電位が高いものが多く、酸化剤としてさらに有効
であることが理解できる (Table 1-1) 5。三重項酸素を直接還元することができるものは強力
な電子供与体に限られるため、生体内で酸素分子の還元を円滑に行うためには反応の活性
化エネルギーを低下させる必要がある。つまり、生体内における酸化酵素・酸素添加酵素
は配位結合により酸素分子を捕捉し、段階的な還元反応により活性酸素種を生成しており、
活性中心に存在する遷移金属元素がその中心的な役割を果たしている。
1.2
銅を含有する機能性タンパク質・酵素 3,6
生体系には活性中心に銅イオンを含む数多くの機能性タンパク質や酵素が存在する。銅
含有タンパク質はその活性中心の構造によりタイプ I~III の 3 つに分類されており、それぞ
れ異なった機能を発現する。タイプ I 銅の分光学的性質は 600 nm 付近に S-から Cu2+への
LMCT によるε = 3000-5000 M-1 cm-1 の大きな吸収帯を持ち、ESR スペクトルにおいて|A//|
~60×10-4 cm-1 と極めて小さい超微細結合定数を示し、通常濃い青色を有することから
ブルー銅と呼ばれる。単核銅中心は、2 つの His 由来のイミダゾール基と、Cys 由来の
チオレート基、Met 由来のチオエーテル基が配位した四面体構造を形成ており、その酸
化還元電位は 200-800 mV (vs. NHE)と高く、Cu(I)状態をとりやすい構造的特徴を有する。
タイプ I 銅は電子移 動タンパク 質の活性中 心に含まれ ており、代 表的な酵素 に
2
Plastocyanin, Azurin, Pseudoazurin, Amicyanin, Stellacyanin, Rusticyanin が挙げられる。タイ
プ II 銅と呼ばれる単核銅中心は、別名、非ブルー銅タンパク質と呼ばれ、タイプ I 銅に
特徴的な非常に強い LMCT を示さず、電子スペクトルにおいて通常の d-d 吸収帯が観測
される。
タイプ II 銅は 2 価銅に一般的な平面構造もしくは平面四角錐構造をしており、
配位子にチオレート基を含んでいないのが特徴である。タイプ II 銅は酸化還元酵素の
活性中心に含まれており、代表的な酸化酵素に Copper Amine Oxidase, Galactose Oxidase
が、酸素添加酵素に Dopamine β-monooxygenase7 (DβM), Peptidylglycine α-hydroxylating
monooxygenase8 (PHM)が、
またその他の酵素では Cu-Zn SOD などの活性中心に含まれる。
タイプ III 銅は複核の銅中心であり、これらは反強磁性相互作用(-2J > 600 cm-1)をしてい
る。タイプ III 銅は酸素運搬タンパク質や酸素添加酵素の活性中心に含まれており、い
ずれの場合も 2 つの銅(I)イオンと酸素分子が反応してサイドオン(µ-η2:η2)型のパーオキ
ソ種が中間体として生成する。代表的な酵素運搬タンパク質は Hemocyanin、酸素添加
酵素には Tyrosinase, Catechol Oxidase が挙げられる。また、活性中心に複数のタイプ I
~III 銅を有するタンパク質をマルチ銅タンパク質と言い、代表的なものに Ascorbate
Oxidase や Nitrite Reductase、また Cytochrome c Oxidase が挙げられる。
このように、活性中心に銅を含有するタンパク質は銅イオン周りの構造的特徴により分
類されており、それぞれ異なった機能を発現している。また、タイプ I 銅ならびにタイプ III
銅中心を有するタンパク質に比べ、タイプ II 銅中心を有するタンパク質の反応中間体や反
応機構に関する知見は少ない。
1.3
DβM, PHM6
DβM7, PHM8は高等動物の脳内または神経系に広く存在する酸素添加酵素であり、それぞ
れ基質であるドーパミン、ペプチド C-末端グリシンの水酸化を行っている。DβM によるド
ーパミンの水酸化ではノルアドレナリンが生成し、PHM によるペプチド C-末端のグリシン
の水酸化生成物は、続く酵素(PAL, peptidylamidoglycolate lyase)の働きによりペプチド末端ア
ミドに変換される(Scheme 1-1)。これらの酵素はいずれも水酸化反応を触媒し、基質に対し
て一原子酸素添加反応を行っている。アミノ酸配列の研究よりこれら酵素の活性中心の構
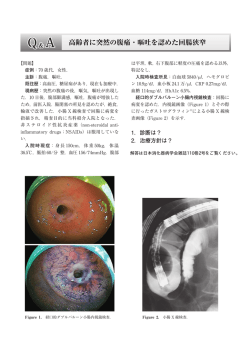
造は非常に近いことが判明している9。PHM は結晶として単離され、その活性中心には 2 つ
の銅イオンが存在することが明らかとなった(Figure 1-1) 10。これら 2 つの銅イオンは CuA,
CuB と名付けられ、CuA は 3 つの His(N)の配位を受けた平面 3 座型の T-shaped 構造であり、
CuB は 2 つの His(N)、1 つの Met(S)、そして H2O(O)の配位を受けた三角錘型の構造であっ
3
た。これら 2 つの銅イオン間の距離は、11Åと遠く離れており、一般的なタイプ III 銅中心
のような複核の銅-酸素種を形成することができない。また近年、PHM より CuB サイトに酸
素分子が end-on 型に配位した結晶も単離されている11。すなわち、これらの 2 つの銅イオン
の役割は明確に分離されており、CuA サイトは電子移動部位、CuB サイトは酸化反応部位と
して機能する12(Figure 1-2)。酸化反応部位である CuB サイトはタイプ II 銅に分類され、単核
の Cu/O2 種を活性種として基質を水酸化すると考えられているが、その反応中間体や反応機
構はわかっていない。
現在、
「単核銅(II)-ハイドロパーオキソ種」もしくは「単核銅(II)-スーパーオキソ種」を反
応中間体とする機構が広く認識されている13。単核銅(II)-ハイドロパーオキソを活性種とす
る機構は、酵素を用いた実験化学的な反応機構の検討から提案された 14 (Scheme 1-2)。
Klinman らは DβM による重水素化ドーパミン、置換フェニルエチルアミンの水酸化実験に
より、反応機構が典型的なラジカル反応であることを明らかとし、18O2 を用いた同位体効果
より、活性種は基質を攻撃する前に O-O 間結合を開裂していることを示唆している15,16,17。
また、このような実験は PHM でも行われており、類似する結果が得られた18。また、スー
パーオキソ種を反応中間体とする機構は銅イオンの酸化状態の異なる二種類の機構が提案
されている 19,20。Amzel らは PHM の結晶の構造学的な解析から、銅(I)-スーパーオキソを活
性種とする機構を提案した19。また、Solomon らは反応機構の計算科学的なシミュレーショ
ンにより単核銅(II)-スーパーオキソを活性種とする機構を提案している20,21。しかしながら、
これらの反応機構を決定づける実験化学的な証拠は存在しない。
単核銅(II)-ハイドロパーオキソ種・単核銅(II)-スーパーオキソ種はともに酸素分子の還元
過程で生成する活性酸素種であるが、酸素分子の還元状態が異なっている。すなわち、酸
素分子の一電子還元によりスーパーオキソ種が、二電子還元により(ハイドロ)パーオキソ種
が生成する。提唱されるどちらの機構も反応過程で単核銅(II)-スーパーオキソ種、さらには
ハイドロパーオキソ種を経由するため、その識別は困難である。すなわち、単核銅(II)-ハイ
ドロパーオキソ種を活性種とする機構は、生成する単核銅(II)-スーパーオキソ種が電子なら
びにプロトンを受け取り単核銅(II)-ハイドロパーオキソ種へと変換されたのち、基質を攻撃
する(Scheme 1-3 top)。これに対し、単核銅(II)-スーパーオキソ種を活性種とする機構は、ス
ーパーオキソ種による基質からの水素原子引き抜きにより単核銅(II)-ハイドロパーオキソ
種が生成し、続く反応が進行する(Scheme 1-3 bottom)。酸素がどの還元段階で基質を攻撃す
るかという点が両機構を区別する鍵となる。
4
1.4
錯体による銅-酸素会合体の合成
DβM, PHM の活性中間体モデルとなり得る、単核銅(II)-スーパーオキソ錯体ならびに単核
銅(II)-ハイドロパーオキソ錯体の合成に関する過去の研究報告と、複核の銅(II)-ハイドロパ
ーオキソ錯体の合成に関する研究報告を以下にまとめた。
単核銅(II)-スーパーオキソ種は酵素の活性中心において銅(I)イオンと酸素分子の反応過
程で常に生成している化学種と考えられている。つまり、酵素や機能性タンパク質のタイ
プ III 銅中心では、酸素分子の一電子還元により単核の銅(II)-スーパーオキソ種が生成し、
そののち早い反応で架橋酸素種を生成していると考えられる。従って、一般的な分光学的
測定により、このような短寿命な反応中間体を観測することは困難である。銅(I)錯体と酸
素分子の反応系においても単核銅(II)-スーパーオキソ種は非常に不安定な化学種であり、多
くの反応系では活性酸素種により架橋された複核錯体が生成する。1990 年代初め Karlin ら
は、このような複核錯体の合成系において反応初期に生成している単核銅(II)-スーパーオキ
ソ錯体の分光学的な観測を行った22。彼らは、配位子に TPA (tris(2-pyridylmethyl)amine)を用
いた銅(I)錯体([CuI(tpa)(RCN)]+)と酸素分子との反応における、単核銅(II)-スーパーオキソ錯
体([CuII(tpa)(O2-)]+)を経由した trans-µ-1,2-peroxo 二核銅錯体([CuII2(tpa)2(O22-)]2+)を合成過程の
観測を行い、その速度論的な解析を報告した(Scheme 1-4)。安定な単核銅(II)-スーパーオキ
ソ錯体の合成は Hydrotris(pyrazolyl)borate を配位子に用いた系において報告されている 23,24。
初期の報告で Thompson らは、hydrotris(3,5-dimethyl-1-pyrazolyl)borate (HB(3,5-Me2(pz))3)を配
位子とした Cu(I)錯体と酸素分子の反応により、析出した赤紫色微粉末が単核銅(II)-スーパ
ーオキソ錯体であることを各種分光学的測定より示唆した23。また 1994 年、Kitajima らは、
嵩高い置換基を導入した hydrotris(pyrazolyl)borate を用いることにより、単核銅(II)-スーパー
オキソ錯体の結晶を単離し、その構造解析に成功している。単離した
[CuII(HB(3-tBu-5-iPrpz)3)(O2-)]錯体は、酸素分子が side-on 型に配位した 5 配位 square pyramidal
型の錯体であり、赤外吸収スペクトル、共鳴ラマンスペクトルによりスーパーオキソ種に
特徴的な吸収帯をそれぞれ 1112/1060 cm-1(16O2/18O2)、
1111/1062 cm-1(16O2/18O2)に観測した24。
また、Tolman らは、嵩高い置換基を有するβ-diketiminate を配位子に用いた系において安定
な単核 Cu/O2 錯体の合成を報告している25。得られた単核 Cu/O2 錯体の結晶は酸素分子が
side-on 型に配位した 4 配位 square planar 型の錯体であり、配位酸素分子間の O-O 間結合長
が 1.392Åであったことや、共鳴ラマンスペクトルにてパーオキソ種に特徴的な吸収帯を
968/917 cm-1(16O2/18O2)に観測したことから、単核銅(III)-パーオキソ錯体であることが判明し
た。一方、嵩高い置換基を有していない hydrotris pyrazolylborate やβ-diketiminate を用いた場
合、Cu(I)錯体と酸素分子の反応によりそれぞれ複核のµ-η2;η2-peroxo dicopper(II)錯体や
5
bis-µ-oxo dicopper(III)錯体を生成することから、単核錯体の合成には嵩高い置換基による複
核化の防止が有効であることが理解できる 26,27 。近年、Schindler や Suzuki らは TREN
(tris(2-aminoethyl)amine)骨格に様々な置換基を有する配位子を用いて、比較的安定な単核銅
(II)-スーパーオキソ錯体を合成している28,29。これらの錯体は、4 座型配位子により銅イオン
周りを配位飽和とすることにより、スーパーオキソイオンが end-on 型に配位していること
が 明 ら か と な っ て い る 。 特 に 嵩 高 い 置 換 基 を 導 入 し た TMG3-TREN
(tris(tetramethylguanidino)tren)を用いたモデル錯体は、一般的に非常に不安定である単核銅
(II)-スーパーオキソ錯体を結晶として単離することに成功しており、得られた結晶はスーパ
ーオキソイオンが end-on 型に配位した 5 配位 trigonal bipyramid 型の錯体であった30(Scheme
1-5)。
銅(II)-ハイドロパーオキソ錯体の合成は、初期には複核錯体の系で報告された。Karlin ら
は配位子に XYL-O- (2,6-bis[bis[2-(2-pyridyl)ethyl]amino]phenolate)を用いた二核の銅(I)錯体を
H+存在下で酸素分子と反応させることにより、µ-1,1 型の架橋ハイドロパーオキソ錯体を合
成した31。前駆体である[CuII2(XYL-O-)(O2-)]+錯体がトリフェニルホスフィンに対し酸化活性
を示さなかったのに対し、[CuII2(XYL-O-)(OOH)]2+錯体はトリフェニルホスフィンに対する
反応性を示し、酸化生成物としてトリフェニルホスフィンオキシドを生成した。近年、Suzuki
らは tris(1-methyl-2-phenyl-4-imidazalylmethyl)amine (L)を配位子とした複核の銅(II)錯体と
過酸化水素の反応により、このようなµ-1,1 型の架橋ハイドロパーオキソ錯体の安定的な合
成と結晶構造を報告している32。[CuII(L)2(OOH)(OH)]2+錯体はヒドロキソ、ハイドロパーオ
キソイオンにより架橋された複核の銅(II)錯体であり、ハイドロパーオキソイオンはイミダ
ゾール窒素との水素結合により安定化された構造を形成していた。また、Kitagawa らは配
位子に bis{2-[N,N-bis(3-pyridylmethyl)-amino]-1,1-dimethylethyl}disulfide (Py2SSPy2)を用いて
合成した複核の銅(I)錯体と過酸化水素の反応により、複核錯体でありながら非架橋型の
end-on ハイドロパーオキソ錯体を合成している33。この錯体は共鳴ラマンスペクトルにおい
て 822 cm-1, 836 cm-1 に二つの O-O 間伸縮振動を観測したことから、配位酸素分子の配位形
式によりシス・トランスの構造異性体が存在することが示唆されている。また、錯体は過
酸化水素を過剰に添加した場合、シクロヘキサンやシクロヘキセンに対する反応性を示す
ことが確認されているが、触媒的な酸化反応は進行しなかった。さらに、このような二核
錯体系での非架橋型ハイドロパーオキソ錯体の合成は、Itoh, Fukuzumi らにより、配位子に
4,5-bis[di(2-pyridylmethyl)aminomethyl]imidazole (Hbdpi)を基本骨格として用いた Cu,Zn-SOD
のモデル錯体でも報告されている34。
単核の銅(II)-ハイドロパーオキソ錯体は主に TPA(tris(2-pyridylmethyl)amine)を基本骨格と
する 5 配位 trigonal bipyramid 型錯体において詳しい検討が行われた。本研究室では嵩高い置
6
換 基 と 水 素 結 合 部 位 を 有 す る 配 位 子 H2BPPA (bis(6-pivalamido-2-pyridylmethy)
(2-pyridylmethyl)amine)を用いた単核銅(II)錯体と過酸化水素の反応により、単核銅(II)-ハイド
ロパーオキソ錯体の結晶化に初めて成功した35(Scheme 1-6)。得られた[CuII(H2bppa)(OOH)]+
錯体は非常に安定な化学種であり、単核銅(II)-ハイドロパーオキソ錯体が示す各種分光学的
特性の解明に大きく貢献した。さらに、TPA を基本骨格として様々な置換基を導入した配
位子を用いることにより、一連の単核銅(II)-ハイドロパーオキソ錯体を合成し、活性種周り
の非共有結合性相互作用がその安定性に及ぼす効果を検討している36。ピリジン 6 位に導入
した様々な置換基により、銅イオン周りの疎水場やハイドロパーオキソイオンに対する水
素結合が、錯体の安定性に及ぼす影響を明らかにし、また、ピリジン 4 位に導入した置換
基による電子的効果が及ぼす影響を検討している。これらの試みにより、錯体に導入した
非共有結合性相互作用部位が不安定中間体の安定化に著しい影響を及ぼすことが明らかと
なった。非 TPA 系の単核銅(II)-ハイドロパーオキソ錯体は、Solomon らが嵩高い置換基を有
す る hydrotris(pyrazolyl)borate を 配 位 子 と し た 合 成 を 報 告 し て い る 37 。 合 成 し た
CuII(HB(3-tBu-5-iPrpz)3)(OOH)錯体は紫外可視吸収スペクトルで~600 nm ( ε = 1540 M-1cm-1)、
~830 nm (ε = ~300 M-1cm-1)に吸収帯を示し、他に報告されている銅(II)-ハイドロパーオキソ
錯体とそのスペクトル的特徴が大きく異なっていた。クメンヒドロペルオキシドを用いて
合成した CuII(HB(3,5-iPr2pz)3)(OOCMe2Ph)錯体は結晶構造が判明しており、そのスペクトル
的特徴の類似性より、CuII(HB(3-tBu-5-iPrpz)3)(OOH)錯体の構造は四面体型であることが示唆
されている。Kodera らは CuB サイトにおける硫黄原子の役割を検討するため38、チオエーテ
ル部位を有する配位子 2-bis(6-methyl-2-pyridylmethylethyl)amino-1-(phenylthio)ethane (N3S)を
用いた単核銅(II)-ハイドロパーオキソ錯体([CuII(N3S)(OOH)]+)を合成した39。ESR スペクトル
の基底状態より、錯体は軸位にチオエーテル硫黄が配位した 5 配位 square pyramid 構造であ
ることが推定され、その分解速度は他の軸配位部位を有する物に比べ著しく安定化してい
た。また、Itoh らは、3 座型の bis[2-(2-pyridyl)ethyl]amine 系の配位子を用いた銅(II)錯体と過
酸化水素の反応において、単核銅(II)-ハイドロパーオキソ錯体を経由した二核銅-酸素錯体
の合成とその速度論的な解析を報告している40。
これらの報告により、モデル錯体を用いた単核銅-酸素錯体の合成と、その分光学的性質
について多くの知見が得られた。しかしながら、これらの錯体は一般的に非常に不安定で
ある単核銅-酸素種を安定的に捕らえることを目的としたものが多く、酸化酵素の反応中間
体モデルであるにも関わらず基質に対する反応性を詳細に検討した例はない。従って、酵
素の反応機構を解明する上で、モデル錯体を用いて合成した銅-酸素種が示す反応性を実験
化学的に検証することが重要と考えられる。
7
1.5
研究の目的
本研究ではタイプ II 銅中心を有する酸素添加酵素、DβM, PHM の反応中間体のモデル化
学的な合成とその物性解析、さらにはその反応性の検証を試みた。現在、主として単核銅(II)スーパーオキソ種、もしくは単核銅(II)-ハイドロパーオキソ種を反応中間体とする機構が提
案されており、これらのモデル化合物は過去に多く合成されているが、実験化学的に基質
に対する詳細な反応性を検討した例はない。従って、提案されている反応機構に対し、モ
デル化合物を用いた実験化学的な裏付けを行うことは極めて重要と考えられる。そこで、
研究は提案されている 2 種類の反応機構に対応させるべく、2 つの戦略に基づいて行った。
ひとつは、「単核銅(II)-スーパーオキソ錯体の合成とその反応性の検討」、 ひとつは「単核
銅(II)-ハイドロパーオキソ錯体の合成とその反応性の検討」である(Scheme 1-7)。
単核銅(II)-スーパーオキソ錯体は通常、単核の銅(I)錯体と酸素分子を反応させることによ
り合成される。過去の報告より、単核銅(II)-スーパーオキソ錯体の合成と安定化には、嵩高
い置換基による金属中心の立体的保護が最も重要と考えられる。このような保護がない場
合、生成する単核銅(II)-スーパーオキソ錯体は自発的に比較的安定な複核の銅-酸素錯体を
形成してしまう。過去に合成された単核銅(II)-スーパーオキソ錯体は、高い安定性ゆえに基
質と反応性を示した例はなく、その原因はこれらの錯体の設計指針に基質との反応に必要
な反応空間が含まれていないためと考えられる。そこで研究では嵩高い置換基を有しなが
らも、基質との反応空間を有した単核銅(II)-スーパーオキソ錯体を合成し、その反応性を検
討した。第 2 章では、配位子に H2BPPA を用いた単核銅(II)-スーパーオキソ錯体が、電子並
びにプロトンを受け取り、単核銅(II)-ハイドロパーオキソ錯体へと変換する反応を追跡した。
さ ら に 第 3 章 で は 、 配 位 子 に BNPA (bis[(6-neopentylamino-2-pyridyl)methyl]
[(2-pyridyl)methyl]amine)を用いた単核銅(II)-スーパーオキソ錯体の外部基質に対する反応性
を検討した。
単核銅(II)-ハイドロパーオキソ錯体は通常、単核銅(II)錯体と過酸化水素を反応させるこ
とにより合成される。単核の銅(II)-ハイドロパーオキソ錯体もまた不安定な化学種であり、
その安定的な合成には嵩高い置換基による活性酸素分子の立体的保護や水素結合によるハ
イドロパーオキソイオンの安定化が有効である。過去に合成された単核銅(II)-ハイドロパー
オキソ錯体の多くは 5 配位型構造であり、これらの錯体は外部基質に対してほとんど反応
性を示さなかった。しかし、酵素の CuB サイトは 4 配位型であることが判明している。そ
こで、研究では 3 座型の配位子 BPBA (N,N-bis (2-pyridylmethyl)-tert-butylamine), BPIPA
(N,N’-Bis(2-pyridylmethyl)iso-propylamine), BPEA (N,N’-Bis(2-pyridylmethyl)-ethylamine)を用い
て 4 配位型の単核銅(II)-ハイドロパーオキソ錯体を合成した。その目的は、外部基質に対す
8
る反応性を発現させること、ならびに配位構造の違いが反応中間体に及ぼす影響を明らか
にすることである。第 4 章では、3 座型配位子 BPBA を用いた単核銅(II)-ハイドロパーオキ
ソ錯体を合成し、過去に報告された 5 配位型錯体との基本的物性や反応性の比較を行った。
また、第 5 章では、配位子 BPIPA, BPEA を用い異なる平面性を有する 4 座型錯体を合成し、
錯体構造がその基本的物性や反応性に与える影響を検討した。
第 6 章では、より高い触媒活性の発現を目的とし、単核鉄(III)-ハイドロパーオキソ錯体
を合成し、各種基質に対する酸化反応を行った。錯体は 4 座型配位子を用い、酸化反応に
優位と考えられるシス位の空配位座を有している。合成した単核鉄(III)-ハイドロパーオキ
ソ錯体の反応活性を、4,5 章で報告した銅錯体と比較した。
第 7 章では、本論文を総括し、低分子量のモデル錯体を用いた金属含有タンパク質の活
性中間体合成の戦略とその有用性についてまとめた。また、異なる 2 つの戦略に基づき行
った「単核銅(II)-スーパーオキソ錯体の合成とその反応性の検討」と「単核銅(II)-ハイドロ
パーオキソ錯体の合成とその反応性の検討」より得られた結果を総合的に判断し、酵素の
反応機構に関する考察を行った。さらには、このようなモデル錯体を用いた触媒の創成の
可能性について言及し、まとめとした。
9
References
1
B. Alberts, D. Bray, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Essential Cell
Biology, Garland Publishing, Inc, New York, 1999
2
化学総説「活性酸素の化学」, 日本化学界編, 学術出版センター, 1990, 7
3
増田秀樹, 福住俊一, 「生物無機化学」, 三共出版, 2005.
4
(a) 松浦輝男, 「酸素酸化反応」, 丸善, 1977. (b) 中野稔, 浅田浩二, 大柳喜彦,「活性酸
素」, 共立出版, 1988.
5
(a) G. Charlot, et al., ed., “Selected Constants of Oxido-Reduction Potentials”, Pergamon Press,
1958. (b) 森永健一, “酸化と還元”, 基礎化学選書, 裳華房, 1972.
6
I. Bertini, A. Sigel and H. Sigel, Handbook on METALLOPROTEINS -Section 15-, Marcel
Dekker, Inc., New York, 2001.
7
(a)N. J. Blackburn, T. M. Pettingill, K. S. Seagraves and R. T. Shigeta, J. Biol. Chem., 1990, 265,
15383-15386. (b) T. M. Pettingill, R. W. Strange and N. J. Blackburn, J. Biol. Chem., 1991, 266,
16996-17003. (c) N. J. Blackburn, In Bioinorganic Chemistry of Copper, K. D. Karlin and Z.
Tyeclar, Eds. Chapman & Hall, New York, 1993, pp 164-183. (d) N. J. Blackburn, F. C. Rhames,
M. Ralle and S. Jaron, J. Biol. Inorg. Chem., 2000, 5, 341-353.
8
(a) D. J. Merkler, R. Kulathila, A. P. Consalvo, S. D. Young and D. E. Ash, Biochemistry, 1992,
31, 7282-7288. (b) B. A. Eipper, A. S. Quon, R. E. Mains, J. S. Boswell and N. J. Blackburn,
Biochemistry, 1995, 34, 2857-2865.
9
(a) P. A. Stoffers, C. B. –R. Green and B. A. Eipper, Proc. Natl. Acad. Sci. USA, 1989, 86,
735-739. (b) A. McMahon, R. Geertman and E. L. Sabban, J. Neurosci. Res., 1990, 25, 395-404.
10 S. T. Prigge, A. S. Kolhekar, B. A. Eipper, R. E. Mains and L. M. Amzel, Science, 1997, 278,
1300-1305.
11 S. T. Prigge, B. A. Eipper, R. E. Mains and L. M. Amzel, Science, 2004, 304, 864-867.
12 J. S. Boswell, B. J. Reedy, R. Kulathila, D. Merkler and N. J. Blackburn, Biochemistry, 1996, 35,
12241-12250.
13 (a) M. H. Gubelmann and A. F. Williams, Struct. Bonding. (Berlin), 1983, 55, 1. (b) K. D. Karlin,
S. Kaderli and A. D. Zuberbuhler, Acc. Chem. Res., 1997, 30, 139.
14 (a) L. C. Stewart and J. P. Klinman, Annu. Rev. Biochem.,1988, 57, 551-592. (b) J. P. Klinman,
Chem. Rev., 1996, 96, 2541-2561.
15 S. M. Miller and J. P. Klinman, Biochemistry, 1998, 22, 3091-3096.
16 S. M. Miller and J. P. Klinman, Biochemistry, 1985, 24, 2114-2127.
17 G. Tian, J. A. Berry and P. Klinman, Biochemistry, 1994, 33, 226-234.
18 (a) W. A. Francisco, D. J. Merkler, N. J. Blackburn and J. P. Klinman, Biochemistry, 1998, 37,
10
8244-8252. (b) K. Takahashi, T. Onami and M. Noguchi, Biochem. J. 1998, 336, 131-137. (c) N.
Ahn and J. P. Klinman, Biochemistry, 1983, 22, 3096-3106. (d) T. M. Zabriskie, H. Cheng and J.
C. Vederas, J. Am. Chem. Soc., 1992, 114, 2270-2272. (e) P. Casera, A. Ganzhorn, C. Phillipo, M.
–C. Chanal and C. Danzin, Bioorg. Med Chem., 1996, 6, 393-396.
19 (a) S. T. Prigge, A. S. Kolkehar, B. A. Eipper, R. E. Mains and L. M. Amzel, Nature Struct. Biol.,
1999, 6, 976-983. (b) S. T. Prigge, R. E. Mains, B. A. Eipper and L. M. Amzel, Cell. Mol. Life.
Sci., 2000, 57, 1236.
20 P. Chen and E. Solomon, J. Am. Chem. Soc., 2004, 126, 4991-5000.
21 J. P. Klinman, J. Biol. Chem., 2006, 281, 3013-3016.
22 (a) K. D. Karlin, N. Wei, B. Jung, S. Kaderli and A. D. Zuberbühler, J. Am. Chem. Soc., 1991,
113, 5868-5870. (b) K. D. Karlin, N. Wei, B. Jung. S. Kaderli, P. Niklaus and A. D. Zuberbühler.,
J. Am. Chem. Soc., 1993, 115, 9506-9514.
23 J. S. Thompson, J. Am. Chem. Soc., 1984, 106, 4057-4059.
24 K. Fujisawa, M. Tanaka, Y. Moro-oka and N. Kitajima, J. Am. Chem. Soc., 1994, 116,
12079-12080.
25 (a) D. J. E. Spencer, N. W. Aboelella, A. M. Reynolds, P. L. Holland and W. B. Tolman, J. Am.
Chem. Soc., 2002, 124, 2108-2109. (b) N. W. Aboelella, S. V. Kryatov, B. F. Gherman, W. W.
Brennessel, V. G. Young. Jr, R. Sarangi, E. V. Rybak-Akimova, K. O. Hodgson, B. Hedman, E. I.
Solomon, C. J. Cramer and W. B. Tolman, J. Am. Chem. Soc., 2004, 126, 16890-16911.
26 N. Kitajima, K. Fujisawa, C. Fujimoto, Y. Moro-oka, S. Hashimoto, T. Kitagawa, K. Toriumi, K.
Tatsumi and A. Nakamura, J. Am. Chem. Soc., 1992, 114, 1277-1291.
27 (a) D. J. E. Spencer, A. M. Reynolds, P. L. Holland, B. A. Jazdzewski, C. Duboc-Toia, L. L.
Pape, S. Yokota, Y. Tachi, S. Itoh and W. B. Tolman, Inorg Chem, 2002, 41, 6307-6321. (b) N. W.
Aboelella, E. A. Lewis, A. M. Reynolds, W. W. Brennessel, C. J. Cramer and W. B. Tolman, J.
Am. Chem. Soc., 2002, 124, 10660-10661.
28 (a) M. Schatz, V. Raab, S. P. Foxon, G. Brehm, S. Schneider, M. Reiher, M. C. Holthausen, J.
Sundermeyer and S. Schindler, Angew. Chem. Int. Ed., 2004, 43, 4360-4363. (b) M. Weitzer and
S. Schindler, Inorg Chem, 2003, 42, 1800-1806. (c) M. Becker, F. W. Heinemann and S.
Schindler, Chem. Eur. J., 1999, 5, 3124-3129. (d) M. Schatz, M. Leibold, S. P. Foxon, M.
Weitzer, F. W. Heinemann, F. Hampel, O. Walter and S. Schindler, DaltonTrans., 2003,
1480-1487.
29 K. Komiyama, H. Furutachi, S. Nagatomo, A. Hashimoto, H. Hayashi, S. Fujinami, M. Suzuki
and T. Kitagawa, Bull. Chem. Soc. Jpn., 2004, 77, 59-72.
30 C. Würtele, E. Gaoutchenova, K. Harms, M. C. Holthausen, J. Sundermeyer and S. Schindler,
Angew. Chem. Int. Ed., 2006, 45, 3867-3869.
11
31 (a) K. D. Karlin, R. W. Cruse and Y. Gultneh, J. Chem. Soc., Chem Commun., 1987, 599-600.
(b) K. D. Karlin, P. Ghosh, R. W. Cruse, A. Farooq, Y. Gultneh, R. R. Jacobson, N. J. Blackburn,
R. W. Strange and J. Zubieta., J. Am. Chem. Soc., 1988, 110, 6769-6780. (c) M. Mahroof-Tahir,
N. N. Murthy, K. D. Karlin, N. J. Blackburn, S. N. Shaikh and J. Zubieta, Inorg Chem, 1992, 31,
3001-3003. (d) D. E. Root, M. Mahroof-Tahir, K. D. Karlin and E. I. Solomon, Inorg Chem,
1998, 37, 4838-4848. (e) L. Q. Hatcher and K. D. Karlin, J. Biol. Inorg. Chem., 2004, 9,
669-683.
32 K. Itoh, H. Hayashi, H. Furutachi, T. Matsumoto, S. Nagatomo, T. Tosha, S. Terada, S. Fujinami,
M. Suzuki and T. Kitagawa, J. Am. Chem. Soc., 2005, 127, 5212-5223.
33 T. Ohta, T. Tachiyama, K. Yoshizawa, T. Yamobe, T. Uchida and T. Kitagawa, Inorg Chem, 2000,
39, 4358-4369.
34 (a) H. Ohtsu, S. Itoh, S. Nagatomo, T. Katagawa, S. Ogo, Y. Watanabe and S. Fukuzumi., Chem.
Commun., 2000, 1051-1052. (b) H. Ohtsu, S. Itoh, S. Nagatomo, T. Katagawa, S. Ogo, Y.
Watanabe and S. Fukuzumi., Inorg Chem, 2001, 40, 3200-3207.
35 A. Wada, M. Harata, K. Hasegawa, K. Jitsukawa, H. Masuda, M. Mukai, T. Kitagawa and H.
Einaga, Angew. Chem. Int. Ed., 1998, 37, 798-800.
36 (a) S. Yamaguchi, S. Nagatomo, T. Kitagawa, Y. Funahashi, T. Ozawa, K. Jitsukawa and H.
Masuda, Inorg Chem, 2003, 42, 6969-6970. (b) S. Yamaguchi, A. Wada, S. Nagatomo, T.
Kitagawa, K. Jitsukawa and H. Masuda, Chem Lett, 2004, 33, 1556-1557. (c) S. Yamaguchi, A.
Kumagai, S. Nagatomo, T. Kitagawa, Y. Funahashi, T. Ozawa, K. Jitsukawa and H. Masuda, Bull.
Chem. Soc. Jpn., 2005, 78, 116-124.
37 P. Chen, K. Fujisawa and E. I. Solomon, J. Am. Chem. Soc., 2000, 122, 10177-10193.
38 B. A. Eipper, A. S. W. Quon, R. E. Mains, J. S. Boswell and N. J. Blackburn, Biochemistry, 1995,
34, 2857-2865.
39 M. Kodera, T. Kita, I. Miura, N. Nakayama, T. Kawata, K. Kano and S. Hirota, J. Am. Chem.
Soc., 2001, 123, 7715-7716.
40 T. Osako, S. Nagatomo, Y. Tachi, T. Kitagawa and S. Itoh, Angew. Chem. Int. Ed., 2002, 41,
4325-4328.
12
Table 1-1 Reductive species of molecular dioxygen
酸性
O2 + e- + H+ → ・OOH
E0 [V]
-0.13
アルカリ性
O2 + e- → O2・-
E0 [V]
-0.563
脱プロトン種
O2・- スーパーオキシド
・OOH + e- + H+ → H2O2
+1.495
O2・- + e- + H2O → -OOH + -OH
+0.413
O22- ペルオキシド
H2O2 + e- + H+ → ・OH + H2O
+0.71
・OH + e- + H+ → H2O
+2.85
-
OOH + e- + H2O → ・OH + 2-OH -0.262
・OH + e- → -OH
O・- 酸素原子アニオンラジカル
O2- オキシド
+2.02
Standard oxidation-reduction potential (in water)
Chemical reaction of DβH
RCH2CH2NH2 (dopamine) + O2 + 2H+ + 2[ascorbate]
→ RCH(OH)CH2NH2 (norepinephrine) + H2O + 2[dehydrosemiascorbate]
Chemical reaction of PHM
RC{O}NHCH2CO2H + O2 + 2H+ + 2[ascorbate]
→ RC{O}NHCH(OH)CO2H + H2O + 2[dehydrosemiascorbate]
Scheme 1-1 Reaction scheme of DβM and PHM.
NH
CuI
HO
HO
N
SH
OH
OH
+
O2, H , e
-
OH
CuII
N
H
NH
O
O
H
CH2NH2
H
O Tyr
II
H
H
Copper(II)-hydroperoxo
Reduced state of CuB site
Cu
H
O
O
CH2NH2
H
O Tyr
H
Homolytic clavage
H+ , e-
OH
OH
norepinephirine
HO
HO
Cu
II
CH2NH2
O
H
CH2NH2
CuII
H
O
H
O Tyr
O Tyr
Radical re-bonding
H-atom abstraction
H
Scheme 1-2 Mechanism for DβM catalysis proposed by Klinman and co-workers.
13
Figure 1-1 Crystal structure of PHM.
(left) Whole structure of PHM.
(right) Local structure of the active site.
Figure 1-2 The structure of the dioxygen binding site.
Dioxygen (the red rod) is shown bound to CuB (the green sphere) in an end-on manner.
14
II
Cu
H+, e-
B-O2
O2, -H2O
H-atom abs.
from substrate
CuIIB-OOH
Substrate is firstly attacked by
hydroperoxo species.
CuIB-OH2
O2, -H2O
H-atom abs.
CuIIB-O2-
Progress of continuring
reaction
CuIIB-OOH
Sub
Sub
Substrate is firstly attacked
by superoxo species.
Scheme 1-3 Difference of the reaction scheme.
Py
CuI NCR
N
Py
Py
Py
O2
CuItpa
-
CuII O2
N
Py
Py
Py
N
Py
CuII O O CuII
Py
Py
Py
Py
N
Scheme 1-4 Observation of mononuclear copper(II)-superoxo species in the generation process of
trans-µ-1,2-peroxo dicopper complex with TPA ligand.
Scheme 1-5 Synthesis of a stable copper(II)-superoxo complex with TMG3-TREN ligand.
15
O
HN
O
NH
N
N
Cu
II
NH
Base, H2O2
O
O
N
NH
N
N
N
O
OH
CuII N
N
Scheme 1-6 Synthesis of stable copper(II)-hydroperoxo species with H2BPPA ligand.
Cu
II
Sub
H2O2, base
II
Cu -OOH
H+ , e -
(Section 4 and 5)
Examination of the reactivity of CuII-OOH
(Section 3)
II
Synthesis of Cu -OOH by the enzymatic reaction process
CuI
O2
ET/PT
CuII-superoxo
ETPT (HAT)
Sub
(Section 2)
Examination of the reactivity of CuII-superoxo
Sub
CuII-OOH
Scheme 1-7 Strategy of this works.
16
第2章
酵素の反応機構に基づいた
単核銅(II)-ハイドロパーオキソ錯体の合成
2.1
序論
1990 年 代 始 め に 、 Karlin ら は [CuI(tpa)(RCN)]+ 錯 体 と 酸 素 分 子 の 反 応 に よ り 複 核 の
trans-µ-1,2-peroxo 錯体([CuII2(tpa)2(O22-)]2+)を合成する過程で、反応のごく初期段階で生成す
る単核銅(II)-スーパーオキソ錯体([CuII(tpa)(O2-)]+)を Stopped flow 法を用いた分光学的測定に
より観測した1。近年、Schindler らは TREN 骨格を有する一連の単核銅(I)錯体と酸素分子の
反応により、安定な単核銅(II)-スーパーオキソ錯体の合成を報告している2。Solomon らは計
算科学的な PHM の反応機構の検討より、単核銅(II)-スーパーオキソ種が基質より水素原子
引き抜きを行なう反応中間体であることを指摘しているが3、過去に合成された単核銅(II)スーパーオキソ錯体が外部基質に対する反応性を示した例はない 2,4。酵素 DβM, PHM の活
性中心では、生成する単核銅(II)-スーパーオキソ種は反応の進行に伴い単核銅(II)-ハイドロ
パーオキソ種に変換されているが、その過程の違いにより異なる反応機構が提案されてい
る5。従って、その変換過程を検討することは反応機構の解明につながる。
本研究では酵素の反応プロセスを模し、錯体分子を用いて合成した単核銅(II)-スーパーオ
キソ錯体の単核銅(II)-ハイドロパーオキソ錯体への変換反応を試みた。一般的に単核銅(II)スーパーオキソ種ならびに単核銅(II)-ハイドロパーオキソ種は不安定な反応中間体であり、
これらの反応を追跡する上で重要なことは化学種の安定化と、錯体の複核化による二核銅酸 素 錯 体 の 生 成 を 防 ぐ こ と と 考 え ら れ る 。 そ こ で 研 究 で は 、 配 位 子 に H2BPPA
(bis(6-pivalamido-2-pyridylmethy)(2-pyridylmethyl)amine)を用いた単核銅(I)錯体を利用した。
H2BPPA はその嵩高い置換基により錯体の複核化を防ぐことが期待される。過去の研究より、
配位子に H2BPPA を用いた銅(II)錯体は、過酸化水素と反応し非常に安定な単核銅(II)-ハイド
ロパーオキソ錯体([CuII(H2bppa)(OOH)]+)を生成することが明らかとなっている6。従って、
この錯体を用いる利点は、単核銅(II)-スーパーオキソ錯体への水素原子の移行反応を、安定
な単核銅(II)-ハイドロパーオキソ錯体の生成をもって確認できる点にある(Scheme 2-1)。本
章では配位子に H2BPPA を用いた単核銅(I)錯体と酸素分子との反応を行ない、単核銅(II)-ス
ーパーオキソ錯体([CuII(H2bppa)(O2-)]+)の合成を行った。また、合成した単核銅(II)-スーパー
オキソ錯体への水素原子の移行反応を、単核銅(II)-ハイドロパーオキソ錯体の生成を観測す
ることにより追跡した。モデル錯体を用いた単核銅(II)-スーパーオキソ錯体の単核銅(II)-ハ
イドロパーオキソ錯体への変換プロセスを実験化学的に再現した例はなく、得られた結果
は酵素の反応機構を明らかにする上で重要な知見となる。
17
2.2
実験
2.2.1
配位子合成
配位子 H2BPPA の構造を Chart 2-1 に示した。配位子は既報の合成法
1
6
により合成し、
H-NMR、元素分析により生成を確認した。
2.2.2
錯体合成
[CuII(Hbppa)]ClO4 錯体は既報の合成法7により合成し、元素分析、ESI-mass スペクトルに
よりその合成を確認した。
2.2.2.1 [CuI(H2bppa)]ClO4 錯体の合成
Ar 雰囲気下において H2BPPA 43.98 mg (0.09 mmol)を溶解させたアセトン溶液 1 ml と、
[CuI(MeCN)4]ClO4 29.43 mg (0.09 mmol)を溶解させたアセトン溶液 1 ml を混合したところ溶
液は赤褐色を呈した。これにジエチルエーテル 15~18 ml を添加し常温にて数時間放置する
ことにより赤色の結晶が析出したため濾取し、乾燥させた。析出した結晶は銅(I)錯体は大
気中で安定であり、元素分析においてその組成の確認を行った。
Yield: 62 %. Anal. Calcd for [CuI(H2bppa)]ClO4・0.5EtOH (C29H36CuClN6O6.5): C, 52.05; H, 5.78;
N, 12.35. Found: C, 52.10; H, 5.52; N, 12.08. 1H-NMR (acetone-d6, 300MHz) ; 1.45(s, 18H,
tert-Bu), 4.09(s, 4H+2H, Mpp+Mp), 7.20(d, J (H-H)= 7.2 Hz, 2H, pp3), 7.49(d, J (H-H)= 7.5 Hz,
1H, p3), 7.54(t, J (H-H)= 6.3 Hz, 1H, p4), 7.77~7.96 (m, 2H+2H+1H, pp4;pp5+p5), 8.73(d, J
(H-H)= 4.5 Hz, 1H, p6), 9.24 (s, 2H, NH).
2.2.3
単核銅(I)錯体と酸素分子との反応
[CuI(H2bppa)]ClO4 錯体と酸素分子との反応は、Ar 雰囲気下において濃度 0.5 mM~1.0 mM
に調製した錯体のアセトンもしくはメタノール溶液に、低温条件下(-40 ~ -80oC)において純
酸素ガスを吹き込むことによって行い、反応に伴う化学的変化を各種分光学的手法により
測定した。また、調製に用いた溶媒はグローブボックスに保存されている脱水溶媒を使用
した。
2.2.4
反応基質
単核銅(II)-スーパーオキソ錯体による水素原子引き抜き反応を確認すべく、TEMPO-H
(2,2,6,6-tetramethyl-1-hydroxypiperidine)並びに Guaiacol (4-methoxyphenol)を基質として用い
た。TEMPO-H のヒドロキシル基は O-H 結合開裂における BDE (bond dissociation energy)が
約 70 kcal mol-1 であり水素を原子として解離し易い化合物である。また、Guaiacol の BDE
は 86.0 kcal mol-1 であるが、こちらも共鳴系によるラジカルの安定化により水素を原子とし
18
て放出しやすい化合物と考えられる。TEMPO-H は既報の合成法を用いて合成し8、Guaiacol
は購入した物を用いた。
2.2.5
測定機器
2.2.5.1
紫外可視吸収(UV-vis)スペクトル
測定装置は、日本分光製 Ubest-V570 紫外可視吸収分光光度計を使用し、測定セルは光路
長 1 cm の石英セルを使用した。サンプルは濃度を 0.25 ~ 1 mM の範囲で調製した錯体溶液
を用い、波長領域 900 ~ 300 nm の範囲において測定を行った。また、極低温での測定につ
いては UNISOKU の低温測定装置を分光器に取り付け、温度制御を行った。
2.2.5.2
電子スピン共鳴(ESR)スペクトル
測定装置は、JEOL JES-RE 1X ESR spectrometer を使用した。サンプルは濃度 1mM に調製
した錯体溶液を市販の ESR サンプルチューブに充填し、液体窒素を満たした測定用デュワ
ーに挿入し凍結させたのち、デュワーごと共振器に取り付けて測定を行った。測定条件を
以下に示した。
Field, 3200±1000 G; Power, 1 mW; Sweep Time, 4 min; Modulation, 0.63 GHz; Time Constant,
0.03 sec.
2.2.5.3
有機微量元素分析
測定装置は、Perkin Elmer 社製 2400II CHNS/O を使用した。試料測定前にガスブランク測
定を行った後、スズカプセルに封入した試料 1.5 ~ 2.0 mg を 2 回測定し、それを元素分析用
アセトアニリド標準試料による補正を行うことで C,H,N の各元素含有量(%)を求めた。
2.2.5.4
核磁気共鳴(1H-NMR)スペクトル
測定装置は、Varian Gemini 200 XL-300 型フーリエ変換核磁気共鳴装置を使用した。ケミ
カルシフトの基準物質として、テトラメチルシラン(TMS)を用いた。内径 5mmφ のサンプル
チューブ内に濃度を約 10 mM に調製した試料溶液について、
積算回数を配位子の場合は 16、
錯体の場合は 128 に設定して δ= -0.2 ~ 9.8 ppm の領域で測定した。
2.2.5.5
X 線結晶構造解析
回折データの測定には一辺が 0.1 ~ 0.3 mm の大きさの単結晶を用い、ガラスファイバー状
に結晶をグリースで固定し、-100 °C で測定した。格子定数は、6° < 2θ < 55°の範囲内の適当
な強度の回折点を用いて、最小二乗法により精密化を行った。
強度測定にはリガク社製 CCD 単結晶自動 X 線構造解析装置を用い、グラファイトで単色
19
化した Mo Kα線を X 線源とし、50 kV, 200 mA により測定した。強度が減衰する場合におい
ては decay correction による強度補正を行った。全反射データに対し Lorentz 因子及び偏光因
子の補正を加えたのち、I ≥ 2.00σ(I)の独立した反射を用いて解析を行った。
構造は重原子法により解析し、差フーリエ合成で得られなかった水素原子の座標は、結
晶水以外のものについては計算から求めた。非水素原子には異方性温度因子を適用し、更
に異常分散による補正、及び吸収補正を実行し、完全マトリックス最小二乗法で精密化を
行った。最小にした関数は、Σw(|F0|–|Fc|)2, w–1 = σ2(F0)である。原子散乱因子は International
Tables for Crystallography Vol.9を参照した。
構造解析、精密化は Crystal Structure 構造解析プログラム10により行い、計算は Windows
2000 をオペレーティングシステムにする市販のパーソナルコンピューターにて行った。
2.2.5.6
共鳴ラマン(rR)スペクトル
測 定 装 置 は 、 Ritsu Oyo Kogaku 社 製 Model MC-100DG spectrophotometer 、 Princeton
Instruments 社製 Model LN/CCD-1100-PB (Charge Coupled Device detector)を使用した。光源は、
Model 2060 Spectra Physics (Kr+)イオンレーザーを用いた。測定は励起波長を 406.7 nm、サン
プル濃度を 10 mM に調製した錯体溶液を高速回転用セルに充填し、低温条件下(-10 ~ -80 °C)
にて測定を行った。
2.2.5.7
ESI-mass スペクトル
測定装置は、Micromass 社製 LCT (ESI-TOF 型)質量分析装置を使用した。錯体の濃度は約
50 µM に調製し、マイクロシリンジを用いて毎秒 600 µl/h の速度で溶液をシリンジポンプに
よって噴霧した。校正は NaI を用いて行い、データは Mass Lynx Ver. 3.5 を用いて Windows NT
ワークステーション上にて処理した。
2.2.5.8
Stopped flow 分光測定
測定装置は、Unisoku 製の Stopped flow rapid scan 分光測定装置 RSP-1000 型を使用した。
モニター光源部はシャッター付き CW Xe アークランプ、分光器にはツェルニィターナー型
回折格子仕様の Unisoku 製 MD200、マルチチャンネル測光部には MOS 型高感度フォトダイ
オードアレイを使用した。+20 ~ -90oC の任意の温度に調節された 1 cm 長の測定セル内にお
いて、自動コック付き 3 液ミキシング装置を用いて反応溶液の混合を行い、極短時間にお
ける UV-vis スペクトル変化を測定した。
20
2.3
結果
2.3.1 [CuI(H2bppa)]+錯体の構造
単離した[CuI(H2bppa)]ClO4 錯体の結晶学的パラメータを Table 2-1 に、主な結合長・結合
角を Table 2-2 に、結晶構造の ORTEP 図を Figure 2-1 に示した。
錯体は銅(I)イオンに対し 3 つのピリジン窒素、1 つの三級アミン窒素、そして 1 つのピバ
ロイルアミド酸素が配位した 5 配位型の構造であった。算出されたτ値は 0.53 であり、錯体
は三方両錐型構造と四角錐型構造の中間的な構造であった11。過去に配位子 H2BPPA を用い
て合成された銅(II)錯体、[CuII(H2bppa)](ClO4)2 の結晶構造より算出したτ値は 0.20 であり、
錯体は四角錐型構造に近い配位構造であった。[CuI(H2bppa)]ClO4 錯体は銅イオンの価数が
CuI (d10)であり錯体構造による配位子場安定化エネルギーに差異がない。従って、平面型を
好む CuII (d9)錯体に比べ配位子がより柔軟に配位して三方両錐型に近づいたと考えられる12。
通常、銅(I)錯体は酸素に対し不安定であり、大気曝露により酸化され銅(II)錯体となる場合
が多いが、得られた赤色結晶は大気中でも安定であった。
2.3.2 [CuI(H2bppa)]+錯体と酸素分子との反応
アセトン溶媒中(-80 oC)における UV-vis スペクトル変化を Figure 2-2 に示した。酸素分子
の添加に伴い溶液は淡黄色から緑青色に変化し、UV-vis スペクトルにおいて 375 nm (ε = 700
M-1cm-1), 628 nm (ε = 190 M-1cm-1), 819 nm (ε = 220 M-1cm-1)に特徴的な吸収帯を観測した。375
nm に見られる吸収帯は一般的な Cu/O2 錯体に観測される活性酸素種から CuII イオンに対す
る LMCT と考えられ、628 nm, 819 nm に見られる吸収帯は CuII の d-d 遷移に由来すると帰属
される。同条件における ESR スペクトルを Figure 2-3 に、また ESI-mass スペクトルを Figure
2-4 に示した。ESR スペクトルは複数の銅(II)種に由来する混合スペクトルであり、各種パ
ラメータの算出は困難であった。ESI-mass スペクトルにおいては m/z = 550.3, 584.3 に主な
ピークを観測した(Figure 2-4 top)。解析により m/z = 550.3 のシグナルは[CuII(Hbppa)]+、m/z =
584.3 のシグナルは[CuII(H2bppa)(OOH)]+と同位体パターンが一致した。このことから、ESR
スペクトルにおいて観測された複数の銅(II)種はこれら二種類の化学種に由来していると考
えられ、[CuI(H2bppa)]ClO4 錯体と酸素分子との反応により単核銅(II)-ハイドロパーオキソ錯
体が生成していることが示唆された。さらに、18O2 を用いて反応を行った場合 m/z = 584.3
に見られたピークは完全に消失し、
m/z = 588.3 に新たなピークが出現した(Figure 2-4 bottom)。
これは[CuII(H2bppa)(18O18OH)]+ 錯体に由来するピークと考えられ、反応により生成する銅
(II)-ハイドロパーオキソ錯体は添加した酸素分子に由来して生成していることが明らかと
なった。
21
メタノールまたはアセトン溶媒中、-80 oC にて[CuI(H2bppa)]ClO4 錯体と酸素分子との反応
を行い、共鳴ラマンスペクトルを測定した。メタノール溶媒中のスペクトルを Figure 2-5 に、
アセトン溶媒中におけるスペクトルを Figure 2-6 に、またこれらスペクトルに見られる特徴
的な伸縮振動を過去に報告した[CuII(H2bppa)(OOH)]+錯体の測定値と共に Table 2-3 に示した。
メタノール溶媒中においては 481 cm-1 並びに 864 cm-1 に特徴的なラマンシグナルを観測した。
前者は CuII-O 間の伸縮振動、後者は O-O 間伸縮振動に由来しており、その値は一般的なパ
ーオキソ種に特徴的な領域に観測された。また、アセトン溶液中において観測された 861
cm-1 の伸縮振動もパーオキソ種に特徴的な O-O 間伸縮振動と帰属される。18O2 を用いた反
応溶液の共鳴ラマンスペクトルはメタノール溶液中では 471 cm-1 (∆ν = 10 cm-1)、並びに 817
cm-1 (∆ν = 47 cm-1)、アセトン溶液中では 814 cm-1 (∆ν = 47 cm-1)に低波数シフトしていた。こ
れらの値は一般的な金属-パーオキソ種に見られる酸素原子の同位体シフトに一致したこと
から、生成した化学種は銅(II)-パーオキソ錯体であり、添加した酸素分子に由来して生成し
ていることを確認した。これらの測定値を過去に[CuII(H2bppa)]2+錯体と過酸化水素との反応
により合成した[CuII(H2bppa)(OOH)]+錯体の測定値と比較したところ完全に一致したことか
ら(誤差 1 cm-1)、[CuI(H2bppa)]+錯体と酸素分子との反応により生成した銅(II)-パーオキソ種
は、 [CuII(H2bppa)(OOH)]+錯体 6 であることが明らかとなった。
2.3.3
銅(II)錯体と過酸化水素の反応系との比較:反応機構の検討
2.3.2 項の実験により、[CuI(H2bppa)]+錯体と酸素分子を反応さると、系中には 2 種類の化
学種、[CuII(Hbppa)]+,錯体と[CuII(H2bppa)(OOH)]+錯体が生成していることが明らかとなった。
そこで、銅(II)錯体と過酸化水素との反応による[CuII(H2bppa)(OOH)]+錯体の合成系とスペク
トル的な比較を行うことにより、反応により生成する化学種の存在比を見積もった。
出発とする銅(II)錯体は[CuII(Hbppa)](ClO4)2 を用いた、これは先の銅(I)錯体と酸素分子と
の反応で生成していると考えられる化学種の一つである。-80 oC、アセトン溶液中で
[CuII(Hbppa)]+ 錯体と過酸化水素の反応を行い、その UV-vis スペクトル変化を測定した
(Figure 2-7)。過酸化水素の添加に伴い溶液は青色から濃緑色に変化し、UV-vis スペクトルに
おいて 380 nm (ε = 1200 M-1cm-1), 648 nm (ε = 205 M-1cm-1), 817 nm (ε = 330 M-1cm-1)に特徴的
な吸収帯を観測した。380 nm に見られる吸収帯は-OOH から CuII に対する特徴的な LMCT
であり、648 nm, 817 nm に見られる吸収帯は CuII に由来する d-d 遷移と帰属されている 6。
反応により生成する化学種は[CuII(H2bppa)(OOH)]+錯体であり、これは 2.3.2 項に示した共鳴
ラマンスペクトルの他、ESR スペクトル、ESI-mass スペクトル、さらには X 線結晶構造解
析により詳細に帰属されている 6。2.3.2 項における銅(I)錯体と酸素分子との反応後の UV-vis
スペクトルは、[CuII(Hbppa)]+錯体と[CuII(H2bppa)(OOH)]+錯体のみのスペクトルを一定の割
22
合で足し合わせた混合スペクトルと考えられる。そこでスペクトルのシミュレーションを
行うことにより、生成系に存在している化学種の割合を求めた。[CuII(Hbppa)]+ 錯体と
[CuII(H2bppa)]+錯体の UV-vis スペクトルを 1 : 1 の割合で足しあわせた合成スペクトルを
Figure 2-7 (c)に示した。このスペクトルは Figure 2-2 (solid line)と良い一致を示したことから、
銅 (I) 錯 体 と 酸 素 分 子 と の 反 応 に よ り 生 成 し た 2 つ の 化 学 種 は 、 [CuII(Hbppa)]+ 錯 体 :
[CuII(H2bppa)(OOH)]+錯体が 1 : 1 の割合で存在していることが明らかとなった。
2.3.4
スピントラップ剤を用いた単核銅(II)-スーパーオキソ錯体の捕捉
[CuI(H2bppa)]ClO4 錯体と酸素分子が反応する場合、最初に銅(I)イオンと酸素分子が接触す
ることにより単核銅(II)-スーパーオキソ錯体が生成していると考えられる。一般的に単核銅
(II)-スーパーオキソ種は非常に不安定な反応中間体であり、本反応ではこのような化学種を
経由して単核銅(II)-ハイドロパーオキソ錯体が生成していると考えられる。反応の UV-vis
スペクトル変化を高時間分解能を有する Stopped flow 法を用いて追跡したが、単核銅(II)-ス
ーパーオキソ錯体に帰属されるスペクトル
1,2
を観測することは出来なかった。これはおも
に[CuI(H2bppa)]ClO4 錯体と酸素分子との反応性が低いことに起因している。そこで、単核銅
(II)-スーパーオキソ錯体生成の間接的な証拠を得るため、スピントラップ剤を用いた反応中
間体の捕捉を試みた。
錯体に対し過剰量(40 当量)の DMPO (5,5-dimethyl-1-pyrroline-N-oxide)を添加したアセトン
またはメタノール溶液中にて、[CuI(H2bppa)]+錯体と酸素分子の反応を行い、各種分光学的
測定を行った。メタノール溶液中、-80 oC における UV-vis スペクトル変化を Figure 2-8 に示
した。酸素分子の添加に伴い溶液は淡黄色から赤紫色に変化し、UV-vis スペクトルにおい
てλmax = 488 nm (ε = 1080 M-1cm-1), 561 nm (ε = 650 M-1cm-1, sh)に特徴的な吸収帯を観測した。
このスペクトル的特徴は DMPO 非添加の系と大きく異なり、その形状は過去に報告されて
いる trans-µ-1,2-peroxo 錯体に近いものであったが、そのモル吸光係数は非常に小さかった13。
アセトン溶液中、同条件にて調製したサンプルの ESI-mass スペクトルを Figure 2-9 に示し
た。m/z = 696.4 に観測されたペアレントピークは[CuII(H2bppa)(O2-)(DMPO)]+の同位体パター
ンと一致したことから、過剰に存在する DMPO が反応により生成する単核銅(II)-スーパー
オキソ錯体([CuII(H2bppa)(O2-)]+)を捕捉し、会合体として系中に存在していることが示唆され
た。また、18O2 を用いた反応を行ったところ、ペアレントピークは m/z = 700.4 に観測され、
同位体シフトが見られたことより、この化学種は添加した酸素分子に由来することが明ら
かとなった。
同条件における ESR スペクトルを測定したところ、silent であった。これは、生成した会
合体が銅(II)イオン上に存在する不対電子と、スーパーオキソに由来する DMPO 上の有機ラ
23
ジカルとの距離が近いため、反強磁性相互作用を起こした結果であると考えられる。この
ように、スピントラップ剤である DMPO を用いることにより、通常のスペクトル測定にお
いては観測が困難である不安定中間体([CuII(H2bppa)(O2-)]+錯体)を捕捉し、それを分光学的に
捕らえることに成功した。
24
2.4
2.4.1
考察
単核銅(II)-スーパーオキソ錯体の合成と観測の試み
実験では、[CuI(H2bppa)]+錯体と酸素分子との反応で生成する単核銅(II)-スーパーオキソ錯
体を直接観測することはできなかった。[CuI(tpa)(RCN)]+錯体と酸素分子の反応では、前駆
体として単核銅(II)-ス ーパーオキソ錯体を生成し、その後、二核の銅-酸素種である
trans-µ-1,2-peroxo 錯体が生成することが報告されている 1。しかしながら、[CuI(H2bppa)]+錯
体と酸素分子の反応性は低く、Stopped flow 法を用いた反応の追跡を試みたが単核銅(II)-ス
ーパーオキソ錯体に特徴的なスペクトルを観測することはできなかった。系中では生成す
る単核銅(II)-スーパーオキソ錯体が非常に早い反応により単核銅(II)-ハイドロパーオキソ錯
体に変換されるため、単核銅(II)スーパーオキソ錯体の観測を困難にしていると考えられる。
[CuI(H2bppa)]+錯体と酸素分子との反応性の低さは錯体の構造的な要因に起因しており、酸
素分子が配位する軸位に配位子のアミド酸素が配位していることがその主な原因と考えら
れる。錯体と酸素分子との反応は銅(I)イオンから酸素分子へ電子の移動を伴って進行する
が、過去に測定された[CuII(H2bppa)]2+錯体のアセトン溶媒中における酸化還元電位は 0.64 V
(vs SHE) 6 であり、またアセトン溶媒中における酸素分子の酸化還元電位は-0.61 V (vs SHE)14
であることから、外圏的な電子移動は進行しないと考えられる15。従って、電子が移動する
ためには酸素分子が銅(I)イオンと接触する必要がある。スーパーオキソイオンが銅(II)イオ
ンに配位することによる安定化、そして段階的な酸素分子の還元を通じた自由エネルギー
の解放が反応の driving force になる。しかしながら酸素分子は電荷的に中性な等核二原子分
子であるため電荷の偏りがなく、正電荷を帯びる銅(I)イオンに対する静電的な求核性は有
していない。従って、アミド酸素が軸配位し、嵩高位置換基を有する[CuI(H2bppa)]+錯体の
中心に存在する銅(I)イオンと、酸素分子との接触は困難であり、その反応性が低いと考え
られる。
単核銅(II)-スーパーオキソ種は単核・複核錯体を問わず、銅(I)錯体と酸素分子の反応や、
DβM, PHM の活性中心において必ず生成する化学種である。そこで、反応系中にスピント
ラップ剤を添加しておくことにより、ラジカル種である単核銅(II)-スーパーオキソ錯体の捕
捉・安定化を試みた。このようなスピントラップ剤を用いたスーパーオキソ錯体の捕捉は
過去に Co(II)錯体による報告が存在する16。過剰量の DMPO 存在下において[CuI(H2bppa)]+
錯体と酸素分子との反応を行い、各種分光学的測定を行なったところ、系中には
[CuII(H2bppa)(O2-)(DMPO)]+錯体が生成していることが明らかとなった。興味深いことに、錯
体からのスーパーオキソの離脱や、O-O 間結合の開裂は観測されず、低温条件下では非常
に安定な会合体を形成していた。残念ながら、会合体の共鳴ラマンスペクトル測定は困難
であったが、その UV-vis スペクトル的特徴より配位した酸素分子はパーオキソ的な性質で
あることが予想される。
25
2.4.2
単核銅(II)-スーパーオキソ錯体を経由した単核銅(II)-ハイドロパーオキソ錯体の合成
I
[Cu (H2bppa)]+錯体と酸素分子の反応を行ったところ生成系では単核銅(II)-ハイドロパー
オ キ ソ 錯 体 ([CuII(H2bppa)(OOH)]+) と 、 配 位 子 が 脱 プ ロ ト ン 化 し た 単 核 銅 (II) 錯 体
反応により発生する単核銅(II)([CuII(Hbppa)]+)が 1:1 の割合で生成していることが判明した。
スーパーオキソ錯体([CuII(H2bppa)(O2-)]+)を経由して、単核銅(II)-ハイドロパーオキソ錯体が
生成したと考えられる。その際、[CuII(H2bppa)(O2-)]+錯体は未反応の[CuI(H2bppa)]+錯体を基
質として反応するため、系中に外部基質を添加することなく反応が進行し、生成系では
[CuII(Hbppa)]+錯体が 50%の収率で観測されたと考えられる(Scheme 2-2)。反応は単核銅(II)スーパーオキソ錯体に基質から水素原子が移行することにより進行するが、その際 2 つの
反応機構が存在する。すなわち直接的な水素原子引き抜き反応である ETPT 機構(ET :
electron-transfer, PT : proton-transfer)と、電子・原子が別々に転移する ET/PT 機構である。こ
れらは反応の前後において生成する化学種は同じであるが本質的には異なる反応である。
すなわち ETPT 機構(もしくは HAT : hydrogen atom transfer 機構)は単核銅(II)-スーパーオキソ
種を活性種とする酵素の反応プロセスに相当し、ET/PT 機構は単核銅(II)-ハイドロパーオキ
ソ種を活性種とする反応プロセスに相当している。従って、単核銅(II)-スーパーオキソ種が
ETPT 機構により水素原子を引き抜く能力の有無が、反応機構判別の鍵となる。
本反応では単核銅(II)-スーパーオキソ錯体の攻撃対象となる基質は未反応の銅(I)錯体で
あ り 、 [CuI(H2bppa)]+ 錯 体 は 反 応 に よ り 電 子 と 配 位 子 の ア ミ ド プ ロ ト ン を 失 っ て
[CuII(H2bppa)]+錯体となる。脱プロトン化の対象となるアミド基はアミド酸素が銅イオンに
配位しているため、一般的なアミドプロトンより塩基性が大きく減少していると考えられ
る。錯体は配位子が銅(I)イオン周りを覆った構造であり、生成する[CuII(H2bppa)(O2-)]+錯体
が未反応の銅(I)錯体に配位した trans-µ-1,2-peroxo 種を形成して電子移動しているとは考え
にくく、またそのような化学種に由来するスペクトル的な証拠は得られなかった。従って、
反応は単核銅(II)-スーパーオキソ錯体による直接的な基質攻撃により進行すると考えられ
る。しかしながら、反応は ETPT、ET/PT 両プロセスでの進行が可能であり本反応系ではそ
の判別をすることは困難であった。以下に可能な反応プロセスを示した。
(i) ETPT 機構
生成する単核銅(II)-スーパーオキソ錯体が未反応の銅(I)錯体のアミドプロトンを直接攻
撃する。水素原子の引きにより単核銅(II)-ハイドロパーオキソ錯体が生成、基質は反応と同
時に Cu(I)イオンから配位子へ電子が移動、[CuII(Hbppa)]+錯体を生成する。
(ii) ET/PT 機構
酸素分子は還元を受けることにより、酸化還元電位がより高電位側にシフトする17。従っ
て生成した単核銅(II)-スーパーオキソ錯体の求電子性は高く、未反応の銅(I)錯体より外圏的
な電子移動が進行し単核銅(II)-パーオキソ錯体が生成したのち、プロトン移動が起こり単核
銅(II)-ハイドロパーオキソ錯体が生成する。
26
系中に過剰の外部基質(Guaiacol)を添加し酸素分子の吹き込みを行ったが、基質からの水
素原子引き抜き反応は観測されなかった。従って、このような基質の存在下においても水
素原子は未反応の銅(I)錯体から供給されており、[CuI(H2bppa)]+錯体は効率の良い水素原子
供給試薬と考えることが出来る。このように、本反応系では生成した単核銅(II)-スーパーオ
キソ錯体([CuII(H2bppa)(O2-)]+)は非常に反応性の高い基質から水素原子を受け取り、単核銅
(II)-ハイドロパーオキソ錯体([CuII(H2bppa)(OOH)]+)に変換されることを明らかにした。
27
2.5
結論
本章では、酵素 DβM, PHM の活性中心において進行している単核銅(II)-スーパーオキソ種
から単核銅(II)-ハイドロパーオキソ種への変換反応を、モデル化学的に再現することにより、
未だ明らかとなっていない反応機構に対し実験化学的な知見を得ることを目的とした。
銅(I)錯体と酸素分子を出発としたハイドロパーオキソ錯体の合成は、過去に Karlin らに
より報告されている18。彼らはフェノレート架橋型の配位子を用いた二核の銅(I)錯体と酸素
分子との反応を行うことにより、架橋η2 型パーオキソ錯体を経由した銅(II)-ハイドロパーオ
キソ錯体が生成することを報告した。この反応では 2 つ存在する銅(I)イオンが酸素分子の
二電子還元を行い、またプロトンソースとして無機酸(HBF4)を用いている。生成するハイド
ロパーオキソイオンは 2 つの銅(II)イオンに架橋し安定化されており、架橋η2 型ハイドロパ
ーオキソ錯体を形成していた。しかしながら、酵素はタイプ II 銅中心にてこのような反応
を行っており、生成する化学種は単核の銅(II)-ハイドロパーオキソ種と考えられる。従って、
反応の厳密なモデル化は単核錯体にて行う必要があった。
研究では、合成した単核銅(I)錯体([CuI(H2bppa)]+)と酸素分子との反応を行なったところ、
単核銅(II)-スーパーオキソ錯体([CuII(H2bppa)(O2-)]+)を経由した単核銅(II)-ハイドロパーオキ
ソ錯体([CuII(H2bppa)(OOH)]+)への変換反応を確認した。その際、反応の水素原子源は未反応
の銅(I)錯体であり、生成系には[CuII(H2bppa)(OOH)]+錯体と[CuII(Hbppa)]+錯体が 1:1 の割合
で生成していることを確認した。このように、単核銅(II)-スーパーオキソ種から単核銅(II)ハイドロパーオキソ種への変換過程を単核錯体上で再現した例はなく、本錯体を用いた単
核銅(II)-ハイドロパーオキソ錯体の生成プロセスは非常に興味深い。また、生成した単核銅
(II)-スーパーオキソ錯体は未反応の銅(I)錯体を外部基質として水素原子を得ている点も特
筆すべきである。水素原子の移動は ETPT もしくは ET/PT 機構で進行すると考えられるが、
本反応系においてその判別をすることは困難であった。
本章においては、一般的に制御が困難である単核銅(II)-スーパーオキソ種を、酵素の反応
機構に則したプロセスにより定量的に単核銅(II)-ハイドロパーオキソ種に変換することに
初めて成功した。また、このような試みには、錯体に導入した疎水場や水素結合等の非共
有結合性相互作用部位による厳密な分子制御が非常に有効であることを確認した。
28
References
1
(a) K. D. Karlin, N. Wei, B. Jung, S. Kaderli and A. D. Zuberbühler, J. Am. Chem. Soc., 1991,
113, 5868-5870. (b) K. D. Karlin, N. Wei, B. Jung. S. Kaderli, P. Niklaus and A. D. Zuberbühler.,
J. Am. Chem. Soc., 1993, 11, 9506-9514.
2
(a) M. Schatz, V. Raab, S. P. Foxon, G. Brehm, S. Schneider, M. Reiher, M. C. Holthausen, J.
Sundermeyer and S. Schindler, Angew. Chem. Int. Ed., 2004, 43, 4360-4363. (b) M. Weitzer and
S. Schindler, Inorg Chem, 2003, 42, 1800-1806. (c) M. Becker, F. W. Heinemann and S.
Schindler, Chem. Eur. J., 1999, 5, 3124-3129. (d) M. Schatz, M. Leibold, S. P. Foxon, M.
Weitzer, F. W. Heinemann, F. Hampel, O. Walter and S. Schindler, DaltonTrans., 2003,
1480-1487. (e) C. Würtele, E. Gaoutchenova, K. Harms, M. C. Holthausen, J. Sundermeyer and
S. Schindler, Angew. Chem. Int. Ed., 2006, 45, 3867-3869.
3
P. Chen and E. Solomon, J. Am. Chem. Soc., 2004, 126, 4991-5000.
4
(a) J. S. Thompson, J. Am. Chem. Soc., 1984, 106, 4057-4059. (b) K. Fujisawa, M. Tanaka, Y.
Moro-oka and N. Kitajima, J. Am. Chem. Soc., 1994, 116, 12079-12080. (c)
K.
Komiyama, H. Furutachi, S. Nagatomo, A. Hashimoto, H. Hayashi, S. Fujinami, M. Suzuki and
T. Kitagawa, Bull. Chem. Soc. Jpn., 2004, 77, 59-72.
5 (a) M. H. Gubelmann and A. F. Williams, Struct. Bonding. (Berlin), 1983, 55, 1. (b) K. D. Karlin,
S. Kaderli and A. D. Zuberbuhler, Acc. Chem. Res., 1997, 30, 139. (c) L. C. Stewart and J. P.
Klinman, Annu. Rev. Biochem.,1988, 57, 551-592. (d) J. P. Klinman, Chem. Rev., 1996, 96,
2541-2561. (e) P. Chen and E. Solomon, J. Am. Chem. Soc., 2004, 126, 4991-5000. (f) J. P.
Klinman, J. Biol. Chem., 2006, 281, 3013-3016.
6
A. Wada, M. Harata, K. Hasegawa, K. Jitsukawa, H. Masuda, M. Mukai, T. Kitagawa and H.
Einaga, Angew. Chem. Int. Ed., 1998, 37, 798-800.
7
M. Harata, K. Hasegawa, K. Jitsukawa, H. Masuda and H. Einaga, Bull. Chem. Soc. Jpn., 1998,
71, 1031-1038.
8
E. A. Mader, A. S. Larsen and J. M. Mayer, J. Am. Chem. Soc., 2004, 26, 8066-8067.
9 International Tables for X-ray Crystallography, Ibers, J. A.; Hamilton, W. C. eds., Kynoch Press,
Birmingham, U. K., 1974, Vol. IV.
10
“Crystal Structure” analysis program (ver 3.7) Produced by Rigaku, 2005.
11 A. W. Addison, T. N. Rao, J. Reedijik, J. Rijin, G. C. Vershoor, J. Chem. Soc. Dalton. Trans.,
1984, 7, 1349-1356.
12 F. A. Cotton, G. Wilkinson, P. L. Gaus, “ BASIC INORGANIC CHEMISTRY 3rd Ed.” John Wiley
& Sons, Inc., New York. 1995
13 L. M. Mirica, X. Ottenwaelder and D. P. Stack, Chem. Rev., 2004, 104, 1013-1045.
14 M. E. Peover and B. S. White, Chem Commun, 1965, 183-184.
15 S. V. Kryatov, S. Taktak, I. V. Korendovych, E. V. Rybak-Akimova, J. Kaizer, S. Torelli, X. Shan.
29
S. Mandal, V. L. MacMurdo, A. M. i. Payeras and L. Que. Jr., Inorg Chem, 2005, 44, 85-99.
16 D. E. Hamilton, R. S. Drago and J. Telser, J. Am. Chem. Soc., 1984, 106, 5353-5355.
17 (a) G. Charlot, et al., ed., “Selected Constants of Oxido-Reduction Potentials”, Pergamon Press,
1958. (b) 森永健一, “酸化と還元”, 基礎化学選書, 裳華房, 1972.
18 (a) L. Q. Hatcher and K. D. Karlin, J. Biol. Inorg. Chem., 2004, 9, 669-683. (b) K. D. Karlin, P.
Ghosh, R. W. Cruse, A. Farooq, Y. Gultneh, R. R. Jacobson, N. J. Blackburn, R. W. Strange and
J. Zubieta, J. Am. Chem. Soc., 1988, 110, 6769-6780. (c) M. M. Tahir, N. N. Murthy, K. D.
Karlin, N. J. Blackburn, S. N. Shaikh and J. Zubieta, Inorg. Chem., 1992, 21, 3001-3003.
30
I
+
O2
H-atom abs
II
[Cu (H2bppa)]
[Cu
(H2bppa)(O2-)]+
[CuII(H2bppa)(OOH)]+
(3)
(2)
(1)
Scheme 2-1 The copper(II)-hydroperoxo generation process in Cu-H2BPPA system.
O
pp5
HN
pp4
N
p3
p4
N
N
p5
pp3
mp
mpp
N
p6
H
N
O
Chart 2-1 Chemical structure of ligand H2bppa.
Figure 2-1 ORTEP drawing of [CuI(H2bppa)]+ with the atom labeling scheme. The thermal
ellipsoids are at the 50% probability level, and the hydrogen atoms were omitted for clarity.
31
Table 2-1 Crystallographic data and refinement parameters for [CuI(H2bppa)]ClO4・0.5EtOH
[CuI(H2bppa)]ClO4・0.5EtOH
C29H36ClCuN6O6.5
671.64
red, block
0.20×0.20×0.10
triclinic
P-1 (#2)
9.973(5)
12.38(3)
14.08(3)
112.72(13)
91.375(9)
98.20(4)
1581(5)
2
1.413
702.00
Mo Kα(λ=0.71070Å)
Rigaku/MSC mercury CCD
-100.0
55.0
Total:12632
5318
427
12.45
0.0597/0.1775
1.001
0.000
Complex
Empieical Formula
Formula Weight
Crystal Color
Crystal Dimensions / mm
Crystal System
Space Group
a/ Å
b/ Å
c/ Å
α / deg
β / deg
γ / deg
Cell Volume / Å3
Z value
Dcalc / gcm-3
F (000)
Radiation
Detectometer
T /℃
2θ max /deg
No.of Reflections Measured
No.of Observations
No.of Variables
Reflection/Parameter Ratio
R / Rw
G.O.F
Max Shift/Error
Table 2-2 Selected bond lengths and angles of [CuI(H2bppa)]ClO4.
Selected bond lengths of [CuI(H2bppa)]+ complex (Å)
Cu(1)-N(1)
2.245(3)
Cu(1)-N(6)
2.062(3)
Cu(1)-N(2)
2.148(3)
Cu(1)-O(1)
2.176(3)
Cu(1)-N(4)
2.008(2)
Selected bond angles of [CuI(H2bppa)]+ complex ( o )
O(1)-Cu(1)-N(1)
158.51(10)
N(1)-Cu(1)-N(4)
80.83(11)
O(1)-Cu(1)-N(2)
84.08(11)
N(1)-Cu(1)-N(6)
78.86(13)
O(1)-Cu(1)-N(4)
117.84(11)
N(2)-Cu(1)-N(4) 118.00(12)
O(1)-Cu(1)-N(6)
95.97(13)
N(2)-Cu(1)-N(6) 104.66(11)
N(1)-Cu(1)-N(2)
77.22(13)
N(4)-Cu(1)-N(6) 126.64(14)
32
1
Absorbance
0.8
0.6
0.4
0.2
0
300
400
500
600
700
800
900
Wavelength / nm
Figure 2-2 UV-vis spectra of [CuI(H2bppa)]ClO4 (1 mM) (dotted line) and
after bubbling O2 to [CuI(H2bppa)]ClO4 (solid line) in acetone at -80 °C.
260
280
300
320
340
360
Magnetic Field / Gauss
Figure 2-3 ESR spectrum of the reaction solution of [CuI(H2bppa)]+ with O2 in MeOH.
33
[CuI(H2bppa)]+ + 16O2
O
550.3
NH
O
NH
N
N
O OH
CuII N
N
O
NH
N
N
CuII
N
N
N
[CuII(H2bppa)(OOH)]+
O
[CuII(Hbppa)]+
584.3
[CuI(H2bppa)]+ + 16O2
584.3
588.3
I
+
18
[Cu (H2bppa)] + O2
Figure 2-4 ESI-mass spectra of the reaction solution of [CuI(H2bppa)]+ with O2 in acetone.
(top) reaction of [CuI(H2bppa)]+ with 16O2
m/z = 550.3 : [CuII(Hbppa)]+, m/z = 584.3 : [CuII(H2bppa)(16O16OH)]+
(bottom) isotope shift of the reaction of [CuI(H2bppa)]+ with 18O2
m/z = 588.3 : [CuII(H2bppa)(18O18OH)]+
34
Complex 1+16O2
864
481
Complex 1+18O2
817
471
Raman shift / cm-1
Figure 2-5 rRaman spectra of the reaction solution of [CuI(H2bppa)]+ (1)with O2 in MeOH.
(top) reaction of [CuI(H2bppa)]+ with 16O2 / ν(Cu-16O) = 481 cm-1, ν(16O-16O) = 861 cm-1
(bottom) reaction of [CuI(H2bppa)]+ with 18O2 / ν(Cu-18O) = 471 cm-1, ν(18O-18O) = 817 cm-1
861
(a)
814
860
(b)
(c)
815
(d)
Raman shift/ cm-1
Figure 2-6 Comparison of rRamam spectra for the reaction of ([CuI(H2bppa)]+ + O2 )
system with that of ( [CuII(Hbppa)]+ + H2O2 ) system.
(a) [CuI(H2bppa)]+ + 16O2 (acetone) / ν(16O-16O) = 861 cm-1
(b) [CuI(H2bppa)]+ + 18O2 (acetone-d6) / ν(18O-18O) = 814 cm-1 (∆ν = 47 cm-1)
(c) [CuII(Hbppa)]+ + H216O2 (acetone) / ν(16O-16O) = 860 cm-1
(d) [CuII(Hbppa)]+ + H218O2 (acetone-d6) / ν(18O-18O) = 815 cm-1 (∆ν = 45 cm-1)
35
Table 2-3 rRaman observation for the O-O stretching mode in cm-1
[CuI(H2bppa)]+ + O2
Reaction
16
acetone
MeOH
O-16O
861
864
18
[CuII(Hbppa)]+ + H2O2
O-18O
814
817
16
O-16O
860
863
18
O-18O
815
817
b
1.2
Absorbance
1
0.8
c
0.6
0.4
0.2
a
0
300
400
500
600
700
800
900
1000
1100
Wavelength [nm]
Figure 2-7 Simulation spectrum of the reaction mixture after reaction of [CuI(H2bppa)]+ with O2.
Spectrum c is the sum of the spectra [CuII(H2bppa)(OOH)]+ (b) and [CuII(Hbppa)]+ (a) which
is good agreement with the spectrum after reaction of [CuI(H2bppa)]+ with O2.It makes
reasonable to consider that the mass balance is about 1:1 (complex [CuII(H2bppa)(OOH)]+ :
complex [CuII(Hbppa)]+) after reaction of [CuI(H2bppa)]+ with O2.
36
1.2
1
Absorbance
0.8
0.6
0.4
0.2
0
300 400 500 600 700 800 900 1000 1100
Wavelength [nm]
Figure 2-8 UV-vis spectral change in the reaction of [CuI(H2bppa)]+ with O2
in the presence of a large amount of DMPO.
(dotted line) [CuI(H2bppa)]+ (1 mM) + DMPO (40 mM) / MeOH
(solid line) O2 bubbling into the above mixture at -80 °C
696.4
CuIH2bppa + 16O2 in DMPO (acetone)
550.3
700.4
CuIH2bppa + 18O2 in DMPO (acetone)
550.3
Figure 2-9 ESI-mass spectra of the reaction solution of [CuI(H2bppa)]+ with O2 in the presence of a
large amount of DMPO. (top) [CuI(H2bppa)]+ +
16
O2, m/z = 550.3 : [CuII(Hbppa)]+, 696.4 :
[CuII(H2bppa)(16O2-)(DMPO)]+. (bottom) [CuI(H2bppa)]+ +
700.4 : [CuII(H2bppa)(18O2-)(DMPO)]+.
37
18
O2, m/z = 550.3 : [CuII(Hbppa)]+,
(1)
O
O
HN
O
NH N
CuI N
N
N
Cu(I)H2BPPA (1)
O2
r.d.s.
O
(4)
O
NH
O
NH N O
II
Cu N
N
N
O
ET/PT or ETPT
Cu(II)-superoxo (2)
NH
OH
NH N O
II
Cu N
N
N
Cu(II)-OOH (3)
Scheme 2-2 A plausible reaction process.
(1) [CuI(H2bppa)]+,
2) [CuII(H2bppa)(O2-)]+,
38
(3) [CuII(H2bppa)(OOH)]+,
(4) [CuII(Hbppa)]+
第3章
単核銅(II)-スーパーオキソ錯体の水素原子引き抜き能の検討
3.1
序論
第 2 章では、[CuI(H2bppa)]+錯体と酸素分子を反応させることにより、単核銅(II)-スーパー
オ キ ソ 錯 体 ([CuII(H2bppa)(O2-)]+) を 経 由 し た 単 核 銅 (II)- ハ イ ド ロ パ ー オ キ ソ 錯 体
([CuII(H2bppa)(OOH)]+)の合成に成功した。反応において単核銅(II)-スーパーオキソ錯体は、
未反応の銅(I)錯体より電子ならびにプロトンを受け取り単核銅(II)-ハイドロパーオキソ錯
体へと変換されたが、その過程が ETPT 機構1または ET/PT 機構2で進行しているかの判別は
できなかった。また系中に外部基質を添加したが、基質からの水素原子引き抜き反応は起
こらず、生成した単核銅(II)-スーパーオキソ錯体の反応性の評価はできなかった。モデル錯
体を用いて合成した反応中間体による各種基質に対する反応性を検証することは本研究の
目的のひとつである。
第 2 章において合成した[CuII(H2bppa)(O2-)]+錯体が外部基質に対する反応性を発現するた
めには以下の改善が求められた。(i) [CuI(H2bppa)]+錯体と酸素分子との反応性の向上。(ii) 分
子内プロトンソースの除去。これらを加味した錯体を用いることにより、合成した単核銅
(II)-スーパーオキソ錯体は外部基質に対する反応性の向上が期待される。そこで、配位子
BNPA (bis[(6-neopentylamino-2-pyridyl)methyl][(2-pyridyl)methyl]amine)4 を用いた単核銅(II)-ス
ーパーオキソ錯体の合成を行い、外部基質からの水素原子引き抜き反応を試みた。BNPA 錯
体は単核錯体の維持に重要な嵩高い置換基を残したまま、H2BPPA 錯体が有していた分子内
アミド酸素を除去することにより酸素分子との反応性が向上する。また、BNPA 錯体には分
子内プロトンソースも存在しないため、外部基質との反応性も向上すると考えられる。
[CuI(bnpa)]+錯体と酸素分子との反応により生成する単核銅(II)-スーパーオキソ錯体の推定
構造を Figure 3-1 に示した。
本章では[CuI(bnpa)]+錯体と酸素分子の反応を行い、生成する銅-酸素錯体の分光学的な追
跡を行った。得られた結果を[CuI(H2bppa)]+錯体を用いた反応系と比較することにより、分
子内アミド部位の有無が酸素分子との反応性に与える影響を検討した。また、反応系中に
過剰量の基質を添加することにより、生成する単核銅(II)-スーパーオキソ錯体と外部基質と
の反応を試みた。様々な BDE (bond dissociation energy)3を持つ基質に対して反応を行うこと
により、実験化学的に合成した単核銅(II)-スーパーオキソ錯体が有する反応性を評価した。
39
3.2
3.2.1
実験
配位子合成
配 位 子 BNPA (bis[(6-neopentylamino-2-pyridyl)methyl][(2-pyridyl)methyl]amine) の 構 造 を
Chart 3-1 に示した。配位子は既報の合成法により合成し4、1H-NMR、元素分析により合成
を確認した。1H-NMR (CDCl3, 300MHz ppm from TMS) ; 0.970 (s, 18H, tert-Bu), 3.02 (d, J
(H-H): 6.0 Hz, 4H, -CH2-tBu), 3.68 (s, 4H, -CH2-Py2), 3.89 (s, 2H, -CH2-Py1), 4.54 (s, 2H, NH),
6.24 (d, J (H-H): 8.7 Hz, 2H, Py2a), 6.89 (d, J (H-H): 7.5 Hz, 2H, Py2c), 7.11 (t, J (H-H): 4.8 Hz, 1H,
Py1b), 7.39 (t, J (H-H): 8.0 Hz, 2H, Py2b), 7.60 (t, J (H-H): 4.8 Hz, 1H, Py1c), 7.67 (d, J (H-H): 7.8
Hz, 1H, Py1d), 8.50 (d, J (H-H): 4.8 Hz, 1H, Py1a). Anal. Calcd for BNPA (C28H40N6): C, 73.00; H,
8.75; N, 18.24. Found: C, 73.00; H, 9.01; N, 17.91.
3.2.2 [CuI(bnpa)]+錯体の合成
等モル量の配位子 BNPA と[CuI(AN)4]SbF6 をアセトン、アセトニトリル、メタノール、THF
等の溶媒中で混合することにより[CuI(bnpa)]+錯体の合成を行った。高濃度の錯体溶液中で
は錯体の不均化反応が進行したため、その単結晶化もしくは粉末結晶としての単離は困難
であった。錯体溶液の ESI-mass スペクトルを測定したところ m/z = 523.4 に単一のピークを
観測し、その同位体パターンが[CuI(bnpa)]+錯体と一致したことから錯体の生成を確認した。
3.2.3
単核銅(I)錯体と酸素分子との反応
単核銅(I)錯体と酸素分子との反応は、Ar 雰囲気下において濃度 0.5 mM に調製した錯体の
アセトン、メタノールまたは THF 溶液に、低温条件下(-40 ~ -80 oC)において純酸素ガスを
吹き込むことによって行い、反応に伴う化学的変化を各種分光学的手法により測定した。
溶液の調製にはグローブボックス内に保存されている脱水溶媒を使用した。
スピントラップ剤による単核銅(II)-スーパーオキソ錯体の捕捉は、錯体に対し 100 当量の
DMPO (5,5-dimethyl-1-pyrroline-N-oxide)を Ar 雰囲気下にて添加したのち酸素分子を吹き込
むことにて行った。また、外部基質からの水素原子引き抜き反応は、錯体に対し 100 当量
の基質を Ar 雰囲気下にて添加したのち酸素分子を吹き込むことにて行った。
3.2.4
外部基質
単核銅(II)-スーパーオキソ錯体の反応性を確認するために、様々な BDE (bond dissociation
energy)を有する有機基質を用いた水素原子引き抜き反応を試みた。以下に用いた基質とそ
の BDE を示した。
40
TEMPO-H (2,2,6,6-tetramethyl-1-hydroxypiperidine) (69.2-71.2 kcal mol-1)5, 1-hydroxypiperidine
(77.0 kcal mol-1), 2,6-di-tert-butylphenol (82.1 kcal mol-1), 2,4-di-tert-butylphenol (85.9 kcal mol-1),
Guaiacol
(4-methoxyphenol)
9,10-dihydroanthracene
(78.0
(86.0
kcal
mol-1),
kcal
mol-1),
thioxantene
1,3-cyclohexanediene
(74.6
(73.2-79.0
kcal
kcal
mol-1),
mol-1),
1,4-cyclohexanediene (73.0-77.0 kcal mol-1), phenylhydrazine (72.6 kcal mol-1)
3.2.5
測定機器
3.2.5.1
紫外可視吸収(UV-vis)スペクトル
測定装置は、日本分光製 Ubest-V570 紫外可視吸収分光光度計を使用し、測定セルは光路
長 1 cm の石英セルを使用した。サンプルは濃度を 0.25 ~ 1 mM の範囲で調製した錯体溶液
を用い、波長領域 900 ~ 300 nm の範囲において測定を行った。また、極低温での測定につ
いては UNISOKU の低温測定装置を分光器に取り付け、温度制御を行った。
3.2.5.2
電子スピン共鳴(ESR)スペクトル
測定装置は、JEOL JES-RE 1X ESR spectrometer を使用した。サンプルは濃度 1mM に調製
した錯体溶液を市販の ESR サンプルチューブに充填し、液体窒素を満たした測定用デュワ
ーに挿入し凍結させたのち、デュワーごと共振器に取り付けて測定を行った。測定条件を
以下に示した。
Field, 3200±1000 G; Power, 1 mW; Sweep Time, 4 min; Modulation, 0.63 GHz; Time Constant,
0.03 sec.
3.2.5.3
有機微量元素分析
測定装置は、Perkin Elmer 社製 2400II CHNS/O を使用した。試料測定前にガスブランク測
定を行った後、スズカプセルに封入した試料 1.5 ~ 2.0 mg を 2 回測定し、それを元素分析用
アセトアニリド標準試料による補正を行うことで C,H,N の各元素含有量(%)を求めた。
3.2.5.4
核磁気共鳴(1H-NMR)スペクトル
測定装置は、varian Gemini 200 XL-300 型フーリエ変換核磁気共鳴装置を使用した。ケミ
カルシフトの基準物質として、テトラメチルシラン(TMS)を用いた。内径 5mmφ のサンプル
チューブ内に濃度を約 10 mM に調製した試料溶液について、
積算回数 16 に設定して δ= -0.2
~ 9.8 ppm の領域で測定した。
41
3.2.5.5
X 線結晶構造解析
回折データの測定には一辺が 0.1 ~ 0.3 mm の大きさの単結晶を用い、ガラスファイバー状
に結晶をグリースで固定し、–100 °C で測定した。格子定数は、6° < 2θ < 55°の範囲内の適
当な強度の回折点を用いて、最小二乗法により精密化を行った。
強度測定にはリガク社製 CCD 単結晶自動 X 線構造解析装置を用い、グラファイトで単色
化した Mo Kα線を X 線源とし、50 kV, 200 mA により測定した。強度が減衰する場合におい
ては decay correction による強度補正を行った。全反射データに対し Lorentz 因子及び偏光因
子の補正を加えたのち、I ≥ 2.00σ(I)の独立した反射を用いて解析を行った。
構造は重原子法により解析し、差フーリエ合成で得られなかった水素原子の座標は、結
晶水以外のものについては計算から求めた。非水素原子には異方性温度因子を適用し、更
に異常分散による補正、及び吸収補正を実行し、完全マトリックス最小二乗法で精密化を
行った。最小にした関数は、Σw(|F0|–|Fc|)2, w–1 = σ2(F0)である。原子散乱因子は International
Tables for Crystallography Vol.6を参照した。
構造解析、精密化は Crystal Structure 構造解析プログラム7により行い、計算は Windows
2000 をオペレーティングシステムにする市販のパーソナルコンピューターにて行った。
3.2.5.6
共鳴ラマン(rR)スペクトル
測 定 装 置 は 、 Ritsu Oyo Kogaku 社 製 Model MC-100DG spectrophotometer 、 Princeton
Instruments 社製 Model LN/CCD-1100-PB (Charge Coupled Device detector)を使用した。光源は、
Model 2060 Spectra Physics (Kr+)イオンレーザーを用いた。測定は励起波長を 406.7 nm、サン
プル濃度を 10 mM に調製した錯体溶液を高速回転用セルに充填し、低温条件下(-10 ~ -80 °C)
にて測定を行った。
3.2.5.7
ESI-mass スペクトル
測定装置は、Micromass 社製 LCT (ESI-TOF 型)質量分析装置を使用した。錯体の濃度は約
50 µM に調製し、マイクロシリンジを用いて毎秒 600 µl/h の速度で溶液をシリンジポンプに
よって噴霧した。校正は NaI を用いて行い、データは Mass Lynx Ver. 3.5 を用いて Windows NT
ワークステーション上にて処理した。
3.2.5.8
Stopped flow 分光測定
測定装置は、
UNISOKU 製の Stopped flow rapid scan 分光測定装置 RSP-1000 型を使用した。
42
モニター光源部はシャッター付き CW Xe アークランプ、分光器にはツェルニィターナー型
回折格子仕様の UNISOKU 製 MD200、マルチチャンネル測光部には MOS 型高感度フォト
ダイオードアレイを使用した。+10 ~ -90oC の任意の温度に調節された 1 cm 長の測定セル
内において、自動コック付き 3 液ミキシング装置を用いて反応溶液の混合を行い、極短時
間における UV-vis スペクトル変化を測定した。
43
3.3
結果
3.3.1 [CuI(bnpa)]+錯体の合成と酸素分子との反応
Ar 雰囲気下にてアセトンもしくはメタノール溶媒に溶かした[CuI(MeCN)4]SbF6 と配位子
BNPA を等モル量混合することにより[CuI(bnpa)]SbF6 の錯体溶液(0.5 mM)を調製した。これ
を密閉式の UV-vis セルに封入し、-80 oC まで冷却したのち酸素分子を吹き込み、その UV-vis
スペクトル変化を観測した(Figure 3-2)。反応に伴い溶液は淡黄色から茶褐色に変化し、アセ
トン溶媒を用いた場合はλmax = 459 nm (ε = 2170 M-1cm-1), 585 nm (sh, ε = 330 M-1cm-1), 821
nm (ε = 154 M-1cm-1)に、メタノール溶媒を用いた場合はλmax = 460 nm (ε = 2190 M-1cm-1), 588
nm (sh, ε = 310 M-1cm-1) , 845 nm (ε = 145 M-1cm-1)に特徴的な吸収帯を観測し、溶媒の違いに
よるスペクトル変化は僅かであった。460 nm 付近の吸収帯は生成した銅-酸素錯体に由来す
る LMCT と考えられ、800 nm 付近の吸収帯は銅(II)イオンに由来する d-d 遷移と帰属される。
LMCT と考えられる吸収帯は過去に報告された単核銅(II)-スーパーオキソ錯体の分光学的
特徴8と一致せず、別種の銅-酸素錯体が生成していると推定される。さらに短波長領域の測
定を行なうためジクロロメタンを溶媒とした測定を試みたが、錯体はジクロロメタン溶媒
中で速やかに分解されたため測定は不可能であった。アセトンもしくはメタノール溶媒中
で生成したこの銅-酸素錯体の可逆的な反応性を確認するため、酸素分子との反応後に数分
間アルゴン置換を行なったが、スペクトルは変化せず可逆的な酸素分子の脱着は観測され
なかった。反応の前後において ESR スペクトルを測定したところ silent であり、生成した
化学種は反磁性であることが判明した。また、反応後の ESI-mass スペクトルを測定したと
こ ろ 、 単 核 の 銅 - 酸 素 錯 体 ([CuII(bnpa)(O2-)]+, [CuII(bnpa)(OOH)]+) 、 複 核 の 銅 - 酸 素 錯 体
([Cu2(bnpa)2(O22-)]2+)に由来するピークは観測されず、m/z = 523.4 に[CuI(bnpa)]+錯体に由来す
るペアレントピークを、m/z = 540.4 に[CuII(bnpa)(OH)]+錯体に由来するピークを観測した。
これらは生成する銅-酸素錯体の分解物であると考えられる。生成した銅-酸素錯体における
酸素分子の還元状態を知るため、共鳴ラマンスペクトルの測定を行なったが、錯体が発す
る蛍光によりラマンシグナルの検出は困難であった。生成した銅-酸素錯体の水素原子引き
抜き能を検討するため、低温条件下において銅-酸素会合体を生成したのち、外部基質とし
て過剰量の TEMPO-H や Guaiacol を添加したがスペクトルに変化は見られず、これらの基
質との反応は確認されなかった。
[CuI(bnpa)]+錯体と酸素分子の反応を Stopped flow 法により測定した。Ar 雰囲気下で濃度
0.5 mM に調製した[CuI(bnpa)]+錯体のアセトン溶液と、室温にて酸素分圧 1 atm に保った酸
素飽和アセトン溶媒を、温度 10 oC に制御した UV-vis セル室内で混合し、そのスペクトル
変化を観測した。セル内温度 10 oC における測定結果を Figure 3-3 に示した。セル内では同
44
体積の反応溶液を混合するため、測定時の測定濃度は 0.25 mM であり、錯体に対し約 5 当
量の酸素分子が存在している9。スペクトルはλmax = 463 nm (ε = 830 M-1cm-1)に極大吸収を観
測し、モル吸光係数は若干小さいが通常の UV-vis スペクトル測定で観測されたものと同じ
化学種が生成していると考えられる。この吸光度の時間変化は一次の速度式に従い、その
見かけの速度定数 kobs は 10.8 s-1 であった。さらに低温条件下での測定も試みたが、そのよ
うな環境では銅(I)錯体と酸素分子との反応性が低下し、有効なスペクトルを得ることがで
きなかった。
3.3.2
スピントラップ剤による反応中間体の検出
3.3.1 項にて [CuI(bnpa)]+錯体と酸素分子との反応を行なったが、単核銅(II)-スーパーオキ
ソ錯体に特徴的なスペクトルは観測されず、また外部基質に対する反応性も確認できなか
った。そこで第 2 章 2.3.4 項と同様、銅(I)錯体と酸素分子との反応により生成する単核銅(II)スーパーオキソ錯体の、スピントラップ剤を用いた捕捉とその分光学的検出を試みた10。
Ar 雰囲気下で調製した[CuI(bnpa)]SbF6 錯体のアセトン溶液(0.5 mM)に、錯体に対し 100
当量の DMPO を添加したのち-80 oC に冷却、酸素分子の吹き込みを行い、その UV-vis スペ
クトル変化を測定した(Figure 3-4)。DMPO の添加による[CuI(bnpa)]+錯体のスペクトル変化
はほとんど見られず、その後の酸素分子の吹き込みにより溶液は淡黄色から赤紫色に変化
し、λmax = 500 nm (ε = 1260 M-1cm-1), 570 nm (sh ε = 680 M-1cm-1)に特徴的な極大吸収を観測し
た。そのスペクトル的特徴は 2.3.4 項で報告した[CuII(H2bppa)(O2-)]+錯体と類似していたこと
から、これらの吸収はパーオキソ的な性質を有した配位酸素分子から銅(II)イオンに対する
LMCT と考えられる。同条件における ESR スペクトルは silent であり、ESI-mass スペクト
ル を 測 定 し た と こ ろ m/z = 668.3 に ペ ア レ ン ト ピ ー ク を 観 測 し 、 こ れ は
[CuII(bnpa)(O2-)(DMPO)]+錯体の同位体パターンと一致した。また、18O2 を用いて同様の実験
を行ったところ、m/z = 672.5 に[CuII(bnpa)(18O2-)(DMPO)]+錯体に由来するペアレントピーク
を観測した。これにより、生成した単核銅(II)-スーパーオキソ錯体はスピントラップ剤によ
って[CuII(bnpa)(O2-)(DMPO)]+錯体として存在していることが明らかとなった。会合体の推定
構造を Figure 3-5 に示した。このように反応のごく初期段階で生成する単核銅(II)-スーパー
オキソ錯体は一般的には非常に不安定であるが、生成時に過剰量のスピントラップ剤が存
在することにより効率よく捕捉・検出ができることが[CuI(bnpa)]+錯体の反応系でも明らか
となった。
3.3.3
基質存在下における銅(I)錯体と酸素分子との反応
3.3.1 項、3.3.2 項の検討により、[CuI(bnpa)]+錯体と酸素分子の反応を行ったところ系中に
45
単核銅(II)-スーパーオキソ種が生成し、その後に不可逆的に基質と反応性を示さない銅-酸
素錯体が生成していることが明らかとなった。一般的に、単核銅(II)-スーパーオキソ錯体は
非常に不安定な反応中間体であり、生成と同時に未反応の銅(I)錯体と反応し架橋パーオキ
ソ錯体を形成して安定化する11。しかしながら、これまでの検討により、系中にスピントラ
ップ剤を添加しておくことにより単核銅(II)-スーパーオキソ錯体の捕捉・安定化が可能であ
り、そのような条件では複核の銅(II)-パーオキソ錯体を生成しないことが明らかとなった。
そこで、あらかじめ系中に基質を添加しておくことにより、生成する単核銅(II)-スーパーオ
キソ錯体による基質からの水素原子引き抜き反応を試みた。
Ar 雰囲気下で調製した[CuI(bnpa)]+錯体のアセトン溶液(0.5 mM)に、錯体に対し 100 当量
の TEMPO-H を添加したのち-80 oC に冷却、酸素分子の吹き込みを行いその UV-vis スペク
トル変化を測定した(Figure 3-6)。TEMPO-H は NO-H 間の結合エネルギーが約 70 kcal mol-1
であり、水素を原子として供給しやすい試薬である 3,5。TEMPO-H の添加による[CuI(bnpa)]+
錯体のスペクトル変化はほとんど見られず、その後の酸素分子の吹き込みにより溶液は淡
黄色から淡緑色に変化した。スペクトルは TEMPO-H 非添加の系と大きく異なり、λmax = 380
nm (ε = 960 M-1cm-1), 684 nm (ε = 125 M-1cm-1)に特徴的な極大吸収を観測した。このスペクト
ルは、過去に報告されている 5 配位三方両錐構造を有する単核銅(II)-ハイドロパーオキソ錯
体([CuII(bnpa)(OOH)]+)に非常に類似しており12、基質との反応が進行し単核の銅(II)-ハイド
ロパーオキソ錯体が生成したことが示唆された。反応溶液の ESI-mass スペクトルを測定し
たところ、m/z = 556.4 にペアレントピークを観測し、これは[CuII(bnpa)(OOH)]+錯体と同位
体パターンが一致した。また、18O2 を用いたところ、m/z = 560.4 に[CuII(bnpa)(18O18OH)]+と
考えられるペアレントピークを観測した。これにより、基質存在下での反応による
[CuII(bnpa)(OOH)]+錯体の生成を確認した。
そこで、配位子に BNPA を有する銅(II)錯体[CuII(bnpa)](ClO4)2 と過酸化水素との反応によ
り、別途合成した単核銅(II)-ハイドロパーオキソ錯体の分光学的特徴と比較を行った13。Ar
雰囲気下で濃度 1 mM に調製した[CuII(bnpa)]2+錯体のアセトン溶液を-40 oC に冷却後、1.2 当
量のトリエチルアミン、10 当量の過酸化水素を添加した場合の UV-vis スペクトル変化を
Figure 3-7 に、また同条件におけるその他の分光学的測定の結果を Table 3-1 に示した。塩基・
過酸化水素の添加により溶液は青色から緑色に変化し、UV-vis スペクトルにおいてλmax =
380 nm (ε = 970 M-1cm-1), 683 nm (ε = 150 M-1cm-1), 850 nm (ε = 230 M-1cm-1)に特徴的な吸収帯
を示した。380 nm に見られる吸収帯はハイドロパーオキソイオンから CuII イオンに対する
LMCT と帰属され、他の 2 つの吸収帯は CuII に由来する d-d 遷移と帰属される。また、反応
生成物は rRaman, ESR, ESI-mass スペクトル等より、単核銅(II)-ハイドロパーオキソ錯体
[CuII(bnpa)(OOH)]+であることを確認した(Table 3-1)。TEMPO-H 存在下における銅(I)錯体と
46
酸素分子との反応により得られた UV-vis スペクトルは(Figure3-6)、銅(II)錯体を出発に合成
した単核銅(II)-ハイドロパーオキソ錯体のスペクトルと良い一致を示した。
3.3.4
単核銅(II)-スーパーオキソ種の水素原子引き抜き能の検討
3.3.3 項にて、[CuI(bnpa)]+錯体と酸素分子との反応により生成する単核銅(II)-スーパーオキ
ソ錯体([CuII(bnpa)(O2-)]+)は、系中に過剰に添加しておいた基質から水素原子を引き抜き、単
核銅(II)-ハイドロパーオキソ錯体([CuII(bnpa)(OOH)]+)となることが明らかとなった。そこで、
TEMPO-H 以外にも様々な BDE (bond dissociation energy)を有する基質を系中に添加してお
くことにより、生成する単核銅(II)-スーパーオキソ錯体の各種基質に対する反応性を検討し
た。
用いた基質を Chart 3-2 にその BDE と共に示し、これら基質との反応による単核銅(II)-ハ
イドロパーオキソ錯体生成の有無を Table 3-2 にまとめた。単核銅(II)-ハイドロパーオキソ錯
体の生成は UV-vis スペクトルにより観測し、主に 380 nm に見られる特徴的な吸収帯の出現
を も っ て 判 断 し た 。 TEMPO-H 同 様 、 比 較 的 弱 い O-H 結 合 を 有 す る 基 質 と し て 、
1-hydroxypiperidine (BDE 77.0 kcal mol-1), 2,6-di-tert-butylphenol (BDE 82.1 kcal mol-1),
2,4-di-tert-butylphenol (BDE 85.9 kcal mol-1), 4-methoxyphenol (BDE 86.0 kcal mol-1)を用いた反
応を試みが、水素原子の引き抜き反応は観測されず、1-hydroxypiperidine を除き 3.3.1 項で見
られた銅-酸素会合体に特徴的な吸収を観測した。1-hydroxypiperidine を基質とした場合のス
ペクトルも銅-酸素会合体に特徴的な吸収が観測されたが、そのモル吸光係数は基質非添加
の系のものより小さかった。また、反応溶液の ESI-mass スペクトル測定を行ったが、単核
銅(II)-ハイドロパーオキソ錯体に由来するピークは観測されなかったことから、水素原子の
引き抜き反応は起こらなかったと考えられる。
弱い C-H 結合を有する基質として thioxantene
(BDE 74.6 kcal mol-1), 9,10-dihydroanthracene (BDE 78.0 kcal mol-1), 1,3-cyclohexadiene (BDE
73.2-79.0 kcal mol-1), 1,4-cyclohexadiene (BDE 73.0-77.0 kcal mol-1)についても反応を試みたが、
UV-vis スペクトルにおいて水素原子引き抜き反応は観測されず、ESI-mass 測定においても
単核銅(II)-ハイドロパーオキソ錯体に由来するシグナルは観測されなかった。弱い N-H 結合
を有する phenylhydrazine (BDE 72.6 kcal mol-1)について同様の検討を行ったところ、酸素分
子の吹き込みによる UV-vis スペクトル変化は基質非添加の系と大きく異なり、λmax = 390 nm
(ε = 2150 M-1 cm-1)に極大吸収を観測した。また、反応後の ESI-mass スペクトルでは m/z =
556.4 に[CuII(bnpa)(OOH)]+と考えられるペアレントピークを観測し、18O2 を用いた場合 m/z =
560.4 に[CuII(bnpa)(18O18OH)]+と考えられるペアレントピークを観測したことから、基質より
水素原子の引き抜き反応が進行し、単核銅(II)-ハイドロパーオキソ錯体が生成したと考えら
れる。
47
3.4
考察
3.4.1 [CuI(bnpa)]+錯体と酸素分子との反応生成物
低温条件下にて[CuI(bnpa)]+錯体と酸素分子を反応させたところ Figure 3-2 に見られるよう
なスペクトル変化を示し、反応による銅-酸素錯体の生成が示唆された。反応後のスペクト
ル的特徴は過去に報告されている単核銅(II)-スーパーオキソ錯体のものと一致しなかった 8。
生成物は共鳴ラマンスペクトル測定が困難であったことから、配位している酸素分子の還
元状態を同定することが出来ず、また ESI-mass スペクトル等による直接的な同定も不可能
であった。しかしながら、ESR サイレントであったことから二核の銅-酸素錯体である可能
性が高く、過去に報告された様々な二核銅-酸素種の分光学的特徴を比較することにより生
成種の同定を試みた14。
BNPA は、TPA (tris(2-pyridylmethy)amine)を基本骨格としてピリジン 6 位より 2 本の
neopentylamino 基を導入した配位子である。配位子 TPA を有する銅(I)錯体([CuI(tpa)(RCN)]+)
と酸素分子との反応では trans-µ-1,2-peroxo 二核銅-酸素錯体([CuII2(tpa)2(O22-)]2+)が生成する
ことを過去に Karlin らが報告している15。[CuII2(tpa)2(O22-)]2+錯体は UV-vis スペクトルにおい
てλmax = 525 nm (ε = 11500 M-1cm-1), 590 nm (ε = 7600 M-1cm-1)に特徴的な吸収帯を示し、
Solomon らの軌道計算によると 525 nm に見られる吸収帯は peroxo (πσ*)→CuII(dσ)への LMCT、
590 nm に見られる吸収帯は peroxo (πν*)→CuII(dσ)への LMCT と帰属されている 16 。
[CuI(bnpa)]+錯体と酸素との反応によるスペクトル(λmax = 460 nm (ε = 2190 M-1cm-1), 588 nm
(sh, ε = 310 M-1cm-1) in acetone)は、[CuII2(tpa)2(O22-)]2+錯体に比べ LMCT と考えられる吸収が
短波長シフトし、モル吸光係数も大きく減少していた。しかしながら、過去に TPA 骨格に
様々な置換基を導入した単核銅(I)錯体と酸素分子との反応を行い、置換基の立体障害や水
素結合が生成する二核銅酸素種の分光学的特徴に及ぼす効果が明らかにされている17。TPA
のピリジン 6 位にアミノ基を導入した一連の配位子を Chart 3-3 に、またそれらの配位子を
用いた銅(I)錯体と酸素分子との反応により生成する trans-µ-1,2-peroxo 錯体の分光学的特徴
を Table 3-3 にまとめた 17(a)。これらの錯体の peroxo (πσ*)→CuII(dσ)と帰属される LMCT はア
ミノ基の数が増えるにつれ短波長シフトし、そのモル吸光係数が減少する傾向が見られる。
LMCT の短波長シフトは、アミノ置換基とパーオキソ酸素との水素結合によるπσ*軌道の安
定化により dx2-y2 とのエネルギー差が開くためと考えられ、モル吸光係数の減少は立体障害
による Cu-O 間の距離の伸張が軌道間の遷移確率を低下させるためと考えられている 17。ま
た、peroxo (πν*)→CuII(dσ)に帰属される LMCT はアミノ基の数が増えるにつれ長波長シフト
する傾向があり、モル吸光係数の減少は立体障害による Cu-O 間の距離の伸張と水素結合に
よるパーオキソ種の回転抑制による影響が大きいと考えられる 17。[CuI(bnpa)]+錯体と酸素分
48
子との反応におけるモル吸光係数を二核錯体として換算するとλmax = 460 nm (ε = 4380
M-1cm-1), 588 nm (sh, ε = 620 M-1cm-1) となり、これらの値は Table 3-3 の配位子 BAPA, TAPA
の示すスペクトル的特徴と良い一致を示した。従って、反応により生成した銅-酸素錯体は
水素結合ならびに嵩高い置換基で覆われた trans-µ-1,2-peroxo 錯体([CuII2(bnpa)2(O22-)]2+)と推
定される。
3.4.2
分子内アミド基が酸素との反応性に与える影響
第 2 章にて[CuI(H2bppa)]+錯体と酸素分子との反応を行ったところ、その反応速度はとて
も遅く、stopped flow 法による測定は困難であった。その原因は銅イオン周りの嵩高い置換
基、また配位アミド酸素により酸素分子との置換反応が阻害されるためと考えられた。そ
こで[CuI(bnpa)]+錯体は分子内アミド酸素を除去することによる酸素分子との反応性の向上
を目的の一つとした。
合成した[CuI(bnpa)]+錯体は Stopped flow 法による反応の追跡が可能(0~10 oC)であり、
[CuI(H2bppa)]+錯体に比べ酸素分子との反応性の向上が認められた。しかしながら、その反
応性は Karlin らの報告にある[CuI(tpa)(RCN)]+錯体と酸素分子との反応に比べ非常に小さく、
Stopped flow 測定において低温条件(-10 oC 以下)での測定は困難であった。また、反応によ
り 単 核 銅 (II)- ス ー パ ー オ キ ソ 錯 体 に 特 徴 的 な ス ペ ク ト ル を 観 測 す る こ と は で き ず 、
trans-µ-1,2-peroxo 錯 体 に 由 来 す る 460 nm の LMCT の 増 加 が 確 認 さ れ た 。 こ れ は
[CuI(H2bppa)]+錯体の場合と同様、律速段階の反応である単核銅(II)-スーパーオキソ錯体の生
成段階に比べ、架橋 peroxo 錯体を生成する次段階の反応が早いため、その観測が困難であ
ったと考えられる(2.4.1 項参)。[CuI(bnpa)]+錯体の場合、反応の進行に伴い二核銅-酸素錯体
である trans-µ-1,2-peroxo 錯体([CuII2(bnpa)2(O22-)]2+)を生成するが、[CuI(H2bppa)]+錯体の場合
このような複核種は生成しなかった。配位子 BNPA は H2BPPA が有していたリジットなア
ミド基がなく、フレキシビリティが増したため、嵩高い置換基が二核構造を防止する程の
分子間反発を保てなかったと考えられる。
3.4.3
単核銅(II)-スーパーオキソ錯体の反応性
[CuI(H2bppa)]+錯体の場合と同様に、スピントラップ剤を用いた単核銅(II)-スーパーオキソ
錯体の捕捉・検出を試みたところ、DMPO の添加により UV-vis スペクトルにおいてλmax = 500
nm (ε = 1260 M-1cm-1), 570 nm (sh ε = 680 M-1cm-1)に特徴的な極大吸収を観測した。そのスペ
クトル的特徴は 2.3.4 項で報告した[CuII(H2bppa)(O2-)(DMPO)]+錯体と非常に類似しており、
ESR, ESI-mass 等の結果とあわせ、生成した単核銅(II)-スーパーオキソ錯体を単核の
[CuII(bnpa)(O2-)(DMPO)]+錯体としての捕捉・安定化に成功したことが判明した。このように
49
[CuI(bnpa)]+錯体と酸素分子との反応系においても、系中にスピントラップ剤を過剰に添加
しておくことで錯体間の接触による trans-µ-1,2-peroxo 錯体の生成を防ぎ、生成する単核銅
(II)-スーパーオキソ錯体を捕捉できることが明らかとなった。そこで、スピントラップ剤に
代わり系中に過剰の基質を添加しておくことにより、生成する単核銅(II)-スーパーオキソ錯
体の水素原子引き抜き能を検討した。
そ の 結 果 、 比 較 的 弱 い 水 素 原 子 結 合 部 位 を 持 つ TEMPO-H (BDE 70 kcal mol-1) と
phenylhydrazine (BDE 72.6 kcal mol-1)からの水素原子引き抜き反応を観測した(Table 3-2)。反
応の進行に伴い、系中には単核銅(II)-ハイドロパーオキソ錯体([CuII(bnpa)(OOH)]+)が生成し、
UV-vis スペクトルにおいて特徴的な吸収を示した。TEMPO-H を基質としての反応させた後
のスペクトルを、別途、銅(II)錯体と過酸化水素を反応させて合成した[CuII(bnpa)(OOH)]+錯
体のスペクトルと比較したところ、両スペクトルがほぼ一致した。このことから、
[CuI(bnpa)]+錯体はほぼ 100%の収率で[CuII(bnpa)(OOH)]+錯体に変換されていることが判明
した。また、phenylhydrazine を基質とした場合、反応により 390 nm に LMCT を観測したが、
そのモル吸光係数は[CuII(bnpa)(OOH)]+錯体が示す値のほぼ 2 倍であった。スーパーオキソ
種による phenylhydrazine の酸化を行った場合、反応生成物は 320 nm に極大吸収を有するこ
とが報告されており18、単核銅(II)-ハイドロパーオキソ錯体が示す LMCT と重なったため、
極大吸収のシフトとモル吸光係数の増加が観測されたと考えられる。また、ESI-mass スペ
クトルにでは、これらの反応で生成した[CuII(bnpa)(OOH)]+錯体の直接的な観測にも成功し
ている。
前章では、水素原子の移動反応が ETPT (HAT)または ET/PT のどちらの機構で進行するか
の判別は困難であった。しかし、本反応系では完全に ETPT (HAT)機構で反応が進行してお
り、DβM, PHM の推定反応機構の一つである単核銅(II)-スーパーオキソ種による基質からの
水素原子引き抜き反応をモデル化学的に再現することに成功した。しかしながらその反応
性は低く、合成した単核銅(II)-スーパーオキソ錯体は、BDE < 72.6 kcal mol-1 の基質を用いた
場合に水素原子を引き抜くことが可能であった。錯体を覆う嵩高い置換基により、基質に
は立体的に反応が困難と考えられるものも含まれているため、一概に BDE のみで活性種の
反応性を評価することはできない。しかしながら、酵素が行っている芳香環α位や末端グリ
シンからの水素原子引き抜き反応に比べ、本反応系において生成した単核銅(II)-スーパーオ
キソ種は非常に低いことが明らかとなった。
50
3.5
結論
本章では、配位子 BNPA を用いて合成した銅(I)錯体と酸素分子の反応を行い、生成する
単核銅(II)-スーパーオキソ種の外部基質への反応性を検討した。[CuI(bnpa)]+錯体と酸素分子
との反応性は[CuI(H2bppa)]+錯体と比べ向上しており、Stopped flow による反応の追跡が可能
であった。これは[CuI(H2bppa)]+錯体の軸位に配位していたアミド酸素が[CuI(bnpa)]+錯体に
は存在しないため、銅(I)イオンと酸素分子との接触が容易になったからと考えられる。し
かしながら、UV-vis スペクトルにおいて単核銅(II)-スーパーオキソ錯体に特徴的な吸収を確
認することはできず、複核の trans-µ-1,2-peroxo 錯体が生成していた。剛直なアミド基を還
元した BNPA 錯体はフレキシビリティが増し、
錯体の複核化を防げなかったと考えられる。
[CuI(H2bppa)]+錯体の場合と同様、スピントラップ剤を用いた単核銅(II)-スーパーオキソ錯体
の捕捉・検出を試みた。その結果、系中にスピントラップ剤を添加しておくことで複核錯
体の生成を防ぎ、[CuII(bnpa)(O2-)(DMPO)]+会合体の検出に成功した。そこで、スピントラッ
プ剤に代わり系中に過剰の基質を添加しておくことにより、生成する単核銅(II)-スーパーオ
キソ錯体の水素原子引き抜き反応を検討した。その結果、錯体は TEMPO-H 等の基質から水
素原子を引き抜き、単核銅(II)-ハイドロパーオキソ錯体を生成することが明らかとなった。
種々の基質に対してこのような検討を行ったところ、合成した単核銅(II)-スーパーオキソ錯
体は BDE < 72.6 kcal mol-1 の基質からの水素原子引き抜くことが明らかとなった(Scheme
3-1)。[CuI(bnpa)]+錯体は分子内にプロトンソースが存在しないことから反応は基質からの直
接的な水素原子引き抜き反応(ETPT 機構)で進行する。すなわち、[CuI(bnpa)]+錯体を用いた
反応系は、提唱されている単核銅(II)-スーパーオキソ種を反応中間体とする反応機構に対応
した単核銅(II)-ハイドロパーオキソ錯体の合成プロセスを実験化学的に再現することに成
功した。このように、モデル錯体を用いて合成した単核銅(II)-スーパーオキソ錯体の外部基
質に対する反応性を検討した例はなく、酵素の反応機構を解明する上で非常に興味深い。
今回、用いた基質には錯体を覆う嵩高い置換基により立体的に反応が困難と予想されるも
のも含まれており、一概に BDE のみでその反応性を評価することはできない。しかしなが
ら、今回合成した単核銅(II)-スーパーオキソ錯体の反応性は非常に低く、錯体は酵素が行っ
ている芳香環α位やアルカンからの水素原子引き抜き反応に対する活性を有していないこ
とが明らかとなった。
51
References
1
ETPT (electron-transfer proton-transfer) often referred PCET (proton-coupled electron-transfer)
or HAT (hydrogen atom abstraction)
2
ET/PT (electron-transfer / proton-transfer)
3
Yu-Ran Luo, HANDBOOK OF BOND DISSOCIATION ENERGIES IN ORGANIC
COMPOUNDS, CRC Press, 2002.
4 (a) S. Yamaguchi. I. Tokairin, Y. Wakita, Y. Funahashi, K. Jitsukawa and H. Masuda, Chem. Lett.,
2003, 32, 406-407. (b) M. harata, K. Hasegawa, K. Jitsukawa, H. Masuda and H. Einaga, Bull.
Chem. Soc. Jpn., 1998, 71, 1031-1038.
5
E. A. Mader, A. S. Larsen and J. M. Mayer, J. Am. Chem. Soc., 2004, 26, 8066-8067.
6 International Tables for X-ray Crystallography, Ibers, J. A.; Hamilton, W. C. eds., Kynoch Press,
Birmingham, U. K., 1974, Vol. IV.
7
“Crystal Structure” analysis program (ver 3.7) Produced by Rigaku, 2005.
8
(a) K. D. Karlin, N. Wei, B. Jung, S. Kaderli and A. D. Zuberbühler, J. Am. Chem. Soc., 1991,
113, 5868-5870. (b) K. D. Karlin, N. Wei, B. Jung. S. Kaderli, P. Niklaus and A. D. Zuberbühler.,
J. Am. Chem. Soc., 1993, 11, 9506-9514. (c) M. Schatz, V. Raab, S. P. Foxon, G. Brehm, S.
Schneider, M. Reiher, M. C. Holthausen, J. Sundermeyer and S. Schindler, Angew. Chem. Int.
Ed., 2004, 43, 4360-4363. (d) M. Weitzer and S. Schindler, Inorg Chem, 2003, 42, 1800-1806.
(e) M. Becker, F. W. Heinemann and S. Schindler, Chem. Eur. J., 1999, 5, 3124-3129. (f) M.
Schatz, M. Leibold, S. P. Foxon, M. Weitzer, F. W. Heinemann, F. Hampel, O. Walter and S.
Schindler, DaltonTrans., 2003, 1480-1487. (g) C. Würtele, E. Gaoutchenova, K. Harms, M. C.
Holthausen, J. Sundermeyer and S. Schindler, Angew. Chem. Int. Ed., 2006, 45, 3867-3869.
9
J. M. Achord and C. L. Hussey, Anal. Chem. 1980, 52, 601-602.
10 D. E. Hamilton, R. S. Drago and J. Telser, J. Am. Chem. Soc., 1984, 106, 5353-5355.
11 (a) R. R. Jacobson, Z. Tyeklar, A. Farooq, K. D. Karlin, S. Liu and J. Zubieta, J. Am. Chem. Soc.,
1988, 110, 3690-3692. (b)
12 (a) A. Wada, M. Harata, K. Hasegawa, K. Jitsukawa, H. Masuda, M. Mukai, T. Kitagawa and H.
Einaga, Angew. Chem. Int. Ed., 1998, 37, 798-800. (b) S. Yamaguchi, S. Nagatomo, T. Kitagawa,
Y. Funahashi, T. Ozawa, K. Jitsukawa and H. Masuda, Inorg Chem, 2003, 42, 6969-6970. (c) S.
Yamaguchi, A. Wada, S. Nagatomo, T. Kitagawa, K. Jitsukawa and H. Masuda, Chem Lett, 2004,
33, 1556-1557. (d) S. Yamaguchi, A. Kumagai, S. Nagatomo, T. Kitagawa, Y. Funahashi, T.
Ozawa, K. Jitsukawa and H. Masuda, Bull. Chem. Soc. Jpn., 2005, 78, 116-124. (e) P. Chen, K.
Fujisawa and E. I. Solomon, J. Am. Chem. Soc., 2000, 122, 10177-10193. (f) M. Kodera, T. Kita,
I. Miura, N. Nakayama, T. Kawata, K. Kano and S. Hirota, J. Am. Chem. Soc., 2001, 123,
7715-7716.
52
13 A. Wada, S. Yamaguchi, K. Jitsukawa and H. Masuda, Angew. Chem. Int. Ed., 2005, 44,
5698-5701.
14 (a) L. M. Milica, X. Ottenwaelder and T. D. P. Stack, Chem. Rev, 2004, 104, 1013-1045. (b) E. A.
Lewis and W. B. Tolman, Chem. Rev., 2004, 104, 1047-1076.
15 (a) Z. Tyeklar, R. R. Jacobson, N, Wei, N. Murthy, J. Zubieta and K. Karlin, J. Am. Chem. Soc.,
1993, 115, 2677. (b) K. D. Karlin, N. Wei, B. Jung. S. Kaderli, P. Niklaus and A. D.
Zuberbühler., J. Am. Chem. Soc., 1993, 115, 9506-9514. (c) R. R. Jacobson, Z. Tyeklar, A.
Farooq, K. D. Karlin, S. Liu and J. Zubieta, J. Am. Chem. Soc., 1988, 110, 3690-3692.
16 (a) M. J. Bladwin, P. K. Ross, J. E. Pate, Z. Tyeklar, K. D. Karlin and E. I. Solomon, J. Am.
Chem. Soc., 1991, 113, 8671. (b) E. I. Solomon, F. Tuczer, D. E. Root, C. A. Brown, Chem. Rev.,
1994, 94, 827.
17 (a) A. Wada, Y. Honda. S. Yamaguchi, S. Nagatomo, T. Kitagawa, K. Jitsukawa and H. Masuda,
Inorg Chem, 2004, 43, 5725-5735. (b) S. Yamaguchi, A. Wada, Y. Funahashi, S. Nagatomo, T.
Kitagawa, K. Jitsukawa and H. Masuda, Eur. J. Inorg. Chem., 2003, 4378-4386.
18
H. P. Misra and I. Fridovich, Biochemistry, 1976, 15, 681-687.
53
O
HN
HN
N
N
N
N
N
N
H
N
N
N
H
N
O
BNPA
H2BPPA
Chart 3-1 Chemical structure of ligand H2BPPA and BNPA.
NH
NH
N
O
O
N
Cu
N
N
Figure 3-1 Estimated structure of mononuclear copper(II)-superoxo complex ([CuII(bnpa)(O2-)]+).
1.2
1
Absorbance
0.8
0.6
0.4
0.2
0
300
400
500
600
700
800
900
Wavelength [nm]
Figure 3-2 UV-vis spectra of [CuI(bnpa)]ClO4 (1 mM) (dotted line) and
after bubbling O2 to
[CuI(bnpa)]ClO4 (solid line) in acetone at -80 °C.
54
0.25
0.2
0.18
0.2
0.16
0.15
0.14
3
ln(Ainf/(Ainf-At))
Absorbance
Absorbance
4
3.5
0.12
0.1
0.1
2.5
2
1.5
1
0.5
0.08
0
0.05
350
0
400
450
500
550
0.06
600
0.05
0.1
0.15
0.2
0.25
0.3
time[s]
0
0.1
Wavelength [nm]
0.2
0.3
0.4
0.5
time [s]
Figure 3-3 Representative spectral changes for reaction of [CuI(bnpa)]+ (0.25 mM) with O2 in
acetone at 10 oC (left). The time courses of the absorbance change at 463 nm (right) and first order
kinetic plots (inset). (The plots were exhibited 15 ms interval.)
0.8
0.7
Absorbance
0.6
0.5
0.4
0.3
0.2
0.1
0
300
400
500
600
700
800
Wavelength [nm]
Figure 3-4 UV-vis spectral change in the reaction of [CuI(bnpa)]+ with O2
in the presence of a large amount of DMPO.
(dotted line) [CuI(bnpa)]+ (0.5 mM) + DMPO (50 mM) / acetone
(solid line) O2 bubbling into the acetone solution at -80 °C
55
NH
NH
N
O
N
NO
O
CuII
N
N
Figure 3-5 Plausible structure for [CuII(bnpa)(O2-)(DMPO)]+ complex.
0.8
0.7
Absorbance
0.6
0.5
0.4
0.3
0.2
0.1
0
300
400
500
600
700
800
Wavelength [nm]
Figure 3-6 UV-vis spectral change in the reaction of [CuI(bnpa)]+ with O2 in
the presence of a large amount of TEMPO-H.
(dotted line) [CuI(bnpa)]+ (0.5 mM) + TEMPO-H (50 mM) / acetone
(solid line) O2 bubbling at -80 °C
56
1.2
1
Absorbance
0.8
0.6
0.4
0.2
0
300
400
500
600
700
800
900
Wavelength [nm]
Figure 3-7 UV-vis spectral change in the reaction of [CuII(bnpa)]2+ with H2O2
in acetone at -40 oC. (dotted line) [CuII(bnpa)]2+ (1 mM) , (solid line) Addition
of Et3N (1.2eq) and H2O2 (10eq) into the solution.
Table 3-1 Spectroscopic properties for [CuII(bnpa)(OOH)]+.
Electronic absorption (λmax/nm , ε/M-1cm-1)
380 (970); LMCT, 683 (150), 850 (230); d-d
ESR
g⊥ = 2.22, g// = 2.00, |A⊥| = 94 G, |A//| = 100 G
Resonance Raman
ν = 847 cm-1(16O-16O), 800 cm-1(18O-18O) <∆ν 47 cm-1>
ESI-mass
m/z = 556 [Cu(bnpa)(16O2H)]+, 560 [Cu(bnpa)(18O2H)]+
57
OH
OH
OH
N
N
OH
OH
tBu
But
OMe
tBu
tBu
TEMPO-H
1-hydroxypiperidine
2,4-di-tert-butylphenol
Guaiacol
2,6-di-tert-butylphenol
H
N NH2
S
1,3-cyclohexadiene
1,4-cyclohexadiene
Thioxantene
9,10-dihydroanthracene
Phenylhydrazine
Chart 3-3 Substrates with small BDE.
Table 3-2 Summary of the H-atom abstraction reaction.
< substrate with weak O-H bond >
BDE
H-atom Abs.
TEMPO-H
69.2-71.2
○
1-hydroxypiperidine
77.0
×
2,6-di-tert-butylphenol
82.1
×
2,4-di-tert-butylphenol
85.9
×
4-methoxyphenol
86.0
×
thioxantene
74.6
×
9,10-dihidoroantracene
78
×
1,4-cyclohexadiene
73.0-77.0
×
1,3-cyclohexadiene
73.2-79.0
×
72.6
○
< substrate with weak C-H bond >
< substrate with weak N-H bond >
phenylhydrazine
58
H 2N
N
N
N
N
N
N
N
H2N
H 2N
N
N
N
N
N
NH2
TPA
MAPA
N
N
NH2
N
BAPA
N
NH2
TAPA
Chart 3-2 A series of tripodal TPA-like ligand which was introduced Amine
group in the 6-position of pyridine.
Table 3-3 UV-vis spectra of trans-µ-1,2 peroxo dicopper [CuII2L2(O22-)]2+ complexes.
LMCT/nm (ε/M-1cm-1)
449 (4620), 684 (400)
474 (6860), 660 (680)
500 (9680), 595 (1820)
525 (11500), 590 (7600)
Ligand
TAPA
BAPA
MAPA
TPA
NH
N
N
O2
X
I
Cu
N
N
Cu(I)BNPA
NH
N
N
Sub
Sub
NH
NH
NH
O
O
II
Cu
NH
N
N
H
Cu(II)BNPA-O2
Scheme 3-1 A plausible reaction process.
59
N
N
O
OH
CuII N
N
Cu(II)BNPA-OOH
60
第4章
平面 4 配位型の型単核銅(II)-ハイドロパーオキソ錯体の
合成と反応性の検討
- 5 配位型錯体との基本的物性ならびに反応性の比較 4.1
序論
DβM, PHM は高等動物の神経系に広く存在する酸化酵素であり、それぞれ基質であるド
ーパミンの水酸化、ペプチド C-末端グリシンの水酸化を行い、有用な生理活性物質の合成
を触媒している1。単核銅(II)-ハイドロパーオキソ種はこれらの酵素において推定される活
性種の一つであり2、過去に多数のモデル錯体が合成されている3。本研究室では、一般的に
不安定である単核銅(II)-ハイドロパーオキソ錯体の結晶化に初めて成功しており、その基本
的物性を明らかにする上で多大な貢献をした4。この[CuII(H2bppa)(OOH)]+錯体は嵩高い置換
基による銅中心の立体的保護、並びにアミド水素とハイドロパーオキソイオンとの水素結
合により、[CuII(tpa)(OOH)]+錯体に比べ活性種が特異的に安定化している(Figure 4-1, Table
4-1)。酵素においては活性中心まわりの立体的効果や非共有結合性相互作用による反応空間
の制御がその高い反応性や基質特異性を発現する上で重要な役割を担っている。そこで、
本研究室では、このような非共有結合性相互作用が銅(II)-ハイドロパーオキソ錯体の性質に
与える影響を明らかにするために、TPA 骨格に種々の置換基を導入した一連の 5 配位型の
単核銅(II)-ハイドロパーオキソ錯体を合成し、その検討を行ってきた5。しかしながら、合
成された 5 配位型の銅(II)-ハイドロパーオキソ錯体の外部基質に対する反応性はごく僅かで
あり、基質に対する反応性を有するモデル錯体の設計が求められた。
PHM の結晶における CuB サイトは 4 配位構造であり6、またその反応過程において 4~5
配位型の構造をとることが示唆されている 7 。そこで実験においては三座型配位子 BPBA
(N,N-bis (2-pyridylmethyl)-tert-butylamine)を用いて 4 配位型の単核銅(II)-ハイドロパーオキソ
錯体の合成を行った。合成した 4 配位型の単核銅(II)-ハイドロパーオキソ錯体を、過去に報
告されている 5 配位型錯体と比較することにより、錯体構造がハイドロパーオキソ種の物
性に及ぼす影響を検討した。また、今回合成したハイドロパーオキソ錯体は本質的に酵素
の活性中心により近い 4 配位構造であることや、配位子の立体障害の低減により銅イオン
周りに開いた空間が保持されることから、基質との反応性の向上が期待される。従って、
合成した単核銅(II)-ハイドロパーオキソ錯体の外部基質に対する反応を行い、その反応性の
評価を試みた。
61
4.2
実験
4.2.1
配位子合成
用いた三座型配位子の構造を Chart 4-1 に示した。配位子 TPA は既報の合成法により合成
し8、1H-NMR、元素分析によりその生成を確認した。
4.2.1.1
N,N’-Bis(2-pyridylmethyl)tert-butylamine (BPBA) の合成
1,4-ジオキサン 150ml に tert-butylamine 0.73 g (1.00×10-2 mol), (2-chloromethyl )pyridine
hydrochloride 4.92 g (3.00×10-2 mol)、蒸留水 20 ml に溶解させた KOH 3.37 g (6.00×10-2 mol)
を加え pH 8~9 の条件下にて還流、TLC (展開溶媒;クロロホルム:メタノール=7:1) で反応
の進行を確認しながら約 2 日間攪拌した。反応の進行に伴い KCl が析出し、反応溶液は褐
色を呈した。濾過により KCl を除去したのち、減圧濃縮により暗褐色の油状混合物を得た。
これをクロロホルムに溶解させ 0.1N HCl aq で中和したのち有機相を分取、飽和食塩水、無
水 MgSO4 を用いて脱水処理をしたのち、減圧濃縮してシリカゲルカラム (溶離液;クロロ
ホルム:メタノール= 100 : 0~5)により精製を行った。得られた 黄褐色の油状物はクロロホル
ム/エーテル混合溶媒中にて結晶化を行い、無色の結晶 0.39 g を得た。Yield 15 %
1
H-NMR
(CDCl3, 300MHz ppm from TMS) ; 1.17(s, 9H, tert-Bu), 3.94(s, 4H, -CH2-), 6.99(t, J (H-H)= 5.0 Hz,
2H, Py-Hb), 7.46~7.50(m, 4H, Py-Hc, Hd), 8.39(d, J (H-H)= 6.0 Hz, 2H, Py-Ha)
4.2.2
錯体合成
4.2.2.1 [CuII(bpba)(MeOH)](ClO4)2 錯体の合成
配位子 BPBA 7.66 mg (3×10-2 mmol)と Cu(ClO4)2·6H2O 11.12 mg (3×10-2 mmol)をメタノー
ル約 0.2 ml にそれぞれ溶解させ、この溶液を混合したところ濃青色の溶液を得た。ジエチ
ルエーテル拡散により結晶化を行い、青色柱状の結晶 14.0 mg を得たので X 線結晶構造解
析による構造同定を行った。
Yield: 85 %. Anal. Calcd. for [CuII(bpba)(H2O)](ClO4)2 (C16H21CuCl2N3O8・2H2O): C, 34.70; H,
4.55; N, 7.59. Found: C, 34.68; H, 4.44; N, 7.44.
4.2.2.2. [CuII(bpba)(MeCN)2](ClO4)2 錯体の合成
配位子 BPBA 7.66 mg (3×10-2 mmol)と Cu(ClO4)2·6H2O 11.12 mg (3×10-2 mmol)をアセト
ニトリル約 0.2 ml にそれぞれ溶解させ、この溶液を混合したところ濃青色の溶液を得た。
ジエチルエーテル拡散により結晶化を行い、青色柱状の結晶 14.9 mg を得たので X 線結晶
62
構造解析による構造同定を行った。
Yield: 83 %. Anal. Calcd. for [CuII(bpba)(H2O)](ClO4)2 (C16H19CuCl2N3O8・H2O): C, 35.86; H, 4.33;
N, 7.84. Found: C, 35.92; H, 4.32; N, 7.83.
4.2.2.3 [CuII(bpba)(H2O)](ClO4)2 錯体の合成
配位子 BPBA 7.66 mg (3×10-2 mmol)と Cu(ClO4)2·6H2O 11.12 mg (3×10-2 mmol)をアセトン
約 0.2 ml にそれぞれ溶解させ、この溶液を混合したところ濃青色の溶液を得た。ジエチル
エーテル拡散により結晶化を行い、青色柱状の結晶 12.5 mg を得たので、X 線結晶構造解析
による構造同定を行った。
Yield: 78 %. Anal. Calcd. for [CuII(bpba)(H2O)](ClO4)2 (C16H21CuCl2N3O8・H2O): C, 35.86; H, 4.33;
N, 7.84. Found: C, 35.81; H, 4.35; N, 7.97.
4.2.2.4 [CuII2(bpba)2(µ-OH)2](ClO4)2 錯体の合成
[CuII(bpba)(H2O)](ClO4)2 錯体 16.07 mg (0.03 mmol)を約 1 ml のアセトニトリルに溶かし、
Ar 雰囲気下でトリエチルアミン 15.3 µl ( 0.036 mmol)を添加した。これを嫌気化において冷
蔵庫中にてジエチルエーテル拡散することにより、青色の結晶 10.8 mg を得たので、X 線結
晶構造解析による構造同定を行った。Yield: 83 %.
4.2.2.5 [CuII(bpba)(N3)]ClO4 錯体の合成
単離した[CuII(bpba)(MeOH)](ClO4)2 錯体 16.5 mg (3×10-2 mmol)をメタノール溶媒 10 ml に溶
かし、これに NaN3 1.95 mg (3×10-2 mmol)を加え超音波をかけながら良く攪拌した。配位子
置換反応の進行に伴い NaN3 は徐々に溶解し、溶液は青色から濃緑色に変化した。減圧濃縮
により得た濃緑色油状物を再度少量のメタノールに溶解し、エーテル拡散を行うことによ
り青色板状の結晶を得たので、X 線結晶構造解析による構造同定を行った。
4.2.2.6 [CuII(bpba)Cl](ClO4)2 錯体の合成
単離した[CuII(bpba)(MeOH)](ClO4)2 錯体 16.5 mg (3×10-2 mmol)をメタノール溶媒 10 ml に
溶かし、これに KCl 2.24 mg (3×10-2 mmol)を加え良く攪拌した。配位子置換反応の進行に
伴い、溶液の青色はやや薄く変化した。減圧濃縮により得た青色油状物を再度少量のメタ
ノールに溶解し、エーテル拡散を行うことにより青色板状の結晶を得たので、X 線結晶構造
解析による構造同定を行った。
63
4.2.3
銅(II)錯体と過酸化水素との反応
0.5 ~ 1.0 mM に調製した銅(II)錯体溶液を Ar 雰囲気下にて低温測定用のセルに封入したの
ち、低温セル室に挿入した。サンプルが任意の温度(10 ~ -80 oC)になるまで十分冷却し、溶
媒にて希釈したトリエチルアミン溶液 50 µl (Et3N, 1.2 当量含有)をシリンジにて添加した後
に、Ar ガスを吹き込むことにより攪拌した。次に、希釈した過酸化水素水 50 µl (H2O2 10 当
量含有) をシリンジにて添加し、Ar ガスを吹き込むことにより銅(II)-ハイドロパーオキソ錯
体の合成を行った。
4.2.4
酸化反応実験
量論的な酸化反応実験は、4.2.3 項に従って合成した錯体溶液に、低温条件下にて基質を
添加することにより行った。反応の進行は銅(II)-ハイドロパーオキソ種に見られる特徴的な
LMCT の減衰をもって確認した。反応終了後、未反応の過酸化水素を除去するため過剰量
のトリフェニルホスフィン(PPh3)を添加し、室温まで徐々に昇温した。反応生成物はガスク
ロマトグラフ測定により定量した。
4.2.5
触媒反応
銅(II)錯体 4 µmol、基質 2 mmol、GC 用の内部標準物質をアセトンもしくはアセトニトリ
ル溶媒 2 ml に溶解させ反応容器に封入し、Ar 置換を行なった。これを ice bath で 0 oC に冷
却後トリエチルアミン 8 µmol、過酸化水素水 0.4 mmol をシリンジにて添加して 15 分間攪拌
することにより反応を行なった。反応終了後、反応溶液約 50 µl を取り出し、過剰量のトリ
フェニルホスフィン(PPh3)を添加したのち室温まで昇温させ、ガスクロマトグラフ測定によ
り反応生成物の定量を行なった。
4.2.6
4.2.6.1
測定装置
紫外可視吸収(UV-vis)スペクトル
測定装置は、日本分光製 Ubest-V570 紫外可視吸収分光光度計を使用し、測定セルは光路
長 1 cm の石英セルを使用した。サンプルは濃度を 0.25 ~ 1 mM の範囲で調製した錯体溶液
を用い、波長領域 900 ~ 300 nm の範囲において測定を行った。また、極低温での測定につ
いては UNISOKU の低温測定装置を分光器に取り付け、温度制御を行った。
4.2.6.2
電子スピン共鳴(ESR)スペクトル
測定装置は、JEOL JES-RE 1X ESR spectrometer を使用した。サンプルは濃度 1mM に調製
64
した錯体溶液を市販の ESR サンプルチューブに充填し、液体窒素を満たした測定用デュワ
ーに挿入し凍結させたのち、デュワーごと共振器に取り付けて測定を行った。測定条件を
以下に示した。
Field, 3200±1000 G; Power, 1 mW; Sweep Time, 4 min; Modulation, 0.63 GHz; Time Constant,
0.03 sec.
4.2.6.3
有機微量元素分析
測定装置は、Perkin Elmer 社製 2400II CHNS/O を使用した。試料測定前にガスブランク測
定を行った後、スズカプセルに封入した試料 1.5 ~ 2.0 mg を 2 回測定し、それを元素分析用
アセトアニリド標準試料による補正を行うことで C,H,N の各元素含有量(%)を求めた。
4.2.6.4
核磁気共鳴(1H-NMR)スペクトル
測定装置は、Varian Gemini 200 XL-300 型フーリエ変換核磁気共鳴装置を使用した。ケミ
カルシフトの基準物質として、テトラメチルシラン(TMS)を用いた。内径 5mmφ のサンプル
チューブ内に濃度を約 10 mM に調製した試料溶液について、積算回数を 16 に設定して δ =
-0.2 ~ 9.8 ppm の領域で測定した。
4.2.6.5
X 線結晶構造解析
回折データの測定には一辺が 0.1 ~ 0.3 mm の大きさの単結晶を用い、ガラスファイバー状
に結晶をグリースで固定し、-100 °C で測定した。格子定数は、6° < 2θ < 55°の範囲内の適当
な強度の回折点を用いて、最小二乗法により精密化を行った。
強度測定にはリガク社製 CCD 単結晶自動 X 線構造解析装置を用い、グラファイトで単色
化した Mo Kα線を X 線源とし、50 kV, 200 mA により測定した。強度が減衰する場合におい
ては decay correction による強度補正を行った。全反射データに対し Lorentz 因子及び偏光因
子の補正を加えたのち、I ≥ 2.00σ(I)の独立した反射を用いて解析を行った。
構造は重原子法により解析し、差フーリエ合成で得られなかった水素原子の座標は、結
晶水以外のものについては計算から求めた。非水素原子には異方性温度因子を適用し、更
に異常分散による補正、及び吸収補正を実行し、完全マトリックス最小二乗法で精密化を
行った。最小にした関数は、Σw(|F0|–|Fc|)2, w–1 = σ2(F0)である。原子散乱因子は International
Tables for Crystallography Vol.9を参照した。
構造解析、精密化は Crystal Structure 構造解析プログラム10により行い、計算は Windows
2000 オペレーティングシステムを搭載する市販のパーソナルコンピューターにて行った。
65
4.2.6.6
共鳴ラマン(rR)スペクトル
測 定 装 置 は 、 Ritsu Oyo Kogaku 社 製 Model MC-100DG spectrophotometer 、 Princeton
Instruments 社製 Model LN/CCD-1100-PB (Charge Coupled Device detector)を使用した。光源は、
Model 2060 Spectra Physics (Kr+)イオンレーザーを用いた。測定は励起波長を 406.7 nm、サン
プル濃度を 10 mM に調製した錯体溶液を高速回転用セルに充填し、低温条件下(-10 ~ -80 °C)
にて測定を行った。
4.2.6.7
ESI-mass スペクトル
測定装置は、Micromass 社製 LCT (ESI-TOF 型)質量分析装置を使用した。錯体の濃度は約
50 µM に調製し、マイクロシリンジを用いて毎秒 600 µl/h の速度で溶液をシリンジポンプに
よって噴霧した。校正は NaI を用いて行い、データは Mass Lynx Ver. 3.5 を用いて Windows NT
ワークステーション上にて処理した。
4.2.6.8
Stopped flow 分光測定
測定装置は、Unisoku 製の Stopped flow rapid scan 分光測定装置 RSP-1000 型を使用した。
モニター光源部はシャッター付き CW Xe アークランプ、分光器にはツェルニィターナー型
回折格子仕様の Unisoku 製 MD200、マルチチャンネル測光部には MOS 型高感度フォトダイ
オードアレイを使用した。+10 ~ -90oC の任意の温度に調節された 1 cm 長の測定セル内に
おいて、自動コック付き 3 液ミキシング装置を用いて反応溶液の混合を行い、極短時間に
おける UV-vis スペクトル変化を測定した
4.2.6.9
ガスクロマトグラフ(GC)測定
測定装置として島津製作所製の GC-8APF を用い、検出器は FID、キャリアーガスには窒
素を用いた。解析は島津製作所製のクロマトパック C-R6A 及びクロマロパック C-R8A を
用い、最小ピーク幅を 5 秒、最小ピーク面積を 100 カウントとし、自動処置によりピーク
の面積を計算した。生成物の定量は内部標準法を用い、予め既知の量の反応生成物と内部
標準物質を用いて測定を行い、その面積比から検量線を作成し、その検量線に従って定量
した。以下にそれぞれの基質を用いた場合における分析条件を挙げる。
・ジメチルスルフィド
カラム:PEG-20M,
Chromosorb WAW DMCS、カラム長:3 m、カラム温度:180 ℃、イ
ンジェクション温度:220 ℃、キャリアーガス一次圧:600 kPa、キャリアーガス二次圧:
170 kPa、水素ガス圧:70 kPa、圧縮空気圧:60 kPa、内部標準物質:1-クロロナフタレン、
(保持時間)ジメチルスルフィド:1.1 min、ジメチルスルフォキシド:4.5 min、ジメチルスル
66
フォン:11.2 min.
・チオアニソール、テトラリン
カラム:PEG-20M,
Chromosorb WAW DMCS、カラム長:3 m、カラム温度:205 ℃、イ
ンジェクション温度:240 ℃、キャリアーガス一次圧:600 kPa、キャリアーガス二次圧:
230 kPa、水素ガス圧:70 kPa、圧縮空気圧:60 kPa、内部標準物質:1-クロロナフタレン、
(保持時間)チオアニソール:2.2 min、フェニルメチルスルフォキシド:12.7 min、ジメチル
スルフォン:24.5 min.
・トルエン、エチルベンゼン、クメン
カラム:PEG-20M,
Chromosorb WAW DMCS、カラム長:3 m、カラム温度:185 ℃、イ
ンジェクション温度:230 ℃、キャリアーガス一次圧:600 kPa、キャリアーガス二次圧:
180 kPa、水素ガス圧:70 kPa、圧縮空気圧:60 kPa、内部標準物質:1,2-ジクロロベンゼン、
(保持時間)トルエン:1.2 min、ベンズアルデヒド:2.8 min、ベンジルアルコール:6.6 min、
エチルベンゼン:1.4 min、アセトフェノン:4.6 min、1‐フェニルエチルアルコール:6.6 min、
クメン:1.4 min、クミルアルコール:5.3 min
・シクロヘキセン
カラム:PEG-20M,
Chromosorb WAW DMCS、カラム長:3 m、カラム温度:110 ℃、イ
ンジェクション温度:220 ℃、キャリアーガス一次圧:600 kPa、キャリアーガス二次圧:
180 kPa、水素ガス圧:70 kPa、圧縮空気圧:60 kPa、内部標準物質:1,2-ジクロロベンゼン、
(保持時間)シクロヘキセン:1.0 min、シクロヘキセンオキシド:3.7 min、シクロヘキセノン:
13.0 min、シクロヘキセノール:14.4 min
67
4.3
結果
4.3.1 [CuII(bpba)(MeOH)]2+, [CuII(bpba)(MeCN)2]2+, [CuII(bpba)(H2O)]2+錯体の構造
メタノール、アセトニトリル、アセトン溶媒中より各錯体の単結晶が得られたため X 線
による結晶構造解析を行なった。結晶学的パラメータを Table 4-2、主な結合長と結合角を
Table 4-3、結晶構造の ORTEP 図を Figure 4-2 に示した。
結晶化に用いた溶媒により各錯体の空配位座にはそれぞれ MeOH, MeCN, H2O 分子が配
位していた。アセトン溶媒中では含まれている水分子がエクアトリアル位に配位している
と考えられる。錯体の元素分析を行なったところ、どれも水分子が配位したものとその組
成が一致した。錯体は大気下に長時間放置しておくことにより、配位していた溶媒分子が
大 気 中 の 水 分 子 と 置 き 換 わ っ た と 考 え ら れ る 。 錯 体 は [CuII(bpba)(MeOH)]2+,
[CuII(bpba)(H2O)]2+が 4 配位型の結晶構造であり、典型的な square planar 構造をしていた。ま
た、結晶状態においてはカウンターアニオンである ClO4-が錯体平面の下方より Cu(II)イオ
ンに接近したパッキング構造をしており、[CuII(bpba)(MeOH)]2+錯体の結晶中での Cu-OClO3
間距離は 2.51~2.71Åであった。[CuII(bpba)(MeCN)2]2+錯体は溶媒であるアセトニトリルを
二分子伴い、5 配位型の結晶として単離された。算出されたτ値11は 0.175 であり、その構造
は trigonal bipyramid より square pyramid に近い構造であった。
4.3.2 [CuII(bpba)(X)]2+錯体溶液の分光学的測定 (X=MeOH, MeCN, H2O)
メタノール、アセトニトリル、アセトン溶媒における UV-vis スペクトルを Figure 4-3 に
示した。スペクトルはどれもλmax = 610 ~ 650 nm の領域に平面型の銅(II)錯体に特徴的な d-d
吸収帯を示したことから、錯体は溶液中では平面構造を形成していると考えられる。d-d 吸
収帯の極大波長はメタノール<アセトン<アセトニトリル溶媒の順に短波長シフトが確認
され、溶媒により空配位座に配位している分子が置換していることが示唆された。アセト
ン溶媒中では先の X 線結晶構造解析、並びに元素分析の結果より溶媒中の水分子が配位し
ていると考えられる。また、アセトニトリル溶媒中のスペクトル的特徴から、結晶状態で
は 5 配位型であった[CuII(bpba)(MeCN)2](ClO4)2 錯体も溶液中では 4 配位型構造であると推定
される。
メタノール、アセトニトリル、アセトン溶媒中で各錯体の ESR スペクトルの測定を行っ
た。各溶媒中における測定結果はどれも典型的な CuII(d9)の square planar 型構造に由来する
スペクトルを示した。ただし、アセトニトリル溶媒を用いた場合、スペクトルはブロード
しており、g 値ならびに|A//|値の算出は困難であった。これはサンプルの凍結状態における
分子の配向が測定に不向きであるためと推定される。メタノール、アセトン溶媒中のスペ
68
クトルより算出した g 値ならびに|A//|値を Table 4-4 にまとめた。平面型銅(II)錯体が示す|A//|
値は、経験的に錯体の構造歪みと相関性があることが報告されている12。各錯体の|A//|値は
平面型の銅(II)錯体としては小さい値であり、錯体は溶液中にて歪んだ平面構造であること
か推定された。
4.3.3 [CuII(bpba)(MeOH)]2+錯体と N3-イオンとの反応
[CuII(bpba)(MeOH)]2+錯体のメタノール溶液に 1 当量の NaN3 を添加し、UV-vis, ESR スペ
クトル測定を行った。錯体濃度 0.2 mM における UV-vis スペクトルを Figure 4-4(left)に、ま
た UV-vis, ESR スペクトル的特徴を Table 4-5 にまとめた。
アジ化ナトリウムの添加により溶液は青色から濃緑色に変化し、UV-vis スペクトルにお
いてλmax = 396 nm (ε = 2870 M-1cm-1), 657 nm (ε = 360 M-1cm-1)に特徴的な吸収帯を確認した。
前者は N3-→CuII に対する LMCT、後者は CuII 錯体に由来する d-d 遷移に帰属される。スペ
クトルは平面型の銅(II)錯体に特徴的であり、溶液中において錯体は平面 4 配位型の
[CuII(bpba)(N3-)]+錯体として存在すると考えられる。ESR スペクトルを測定したところ平面
型の銅(II)錯体に特徴的な形状であり、g⊥=2.05, g//=2.22, |A//|=177 であった。また、|A//|値は
錯体が高い平面性を保持していることを示しており、UV-vis による推定構造を支持してい
た。アザイドイオンが配位した錯体は単結晶が得られたため X 線による結晶構造解析を行
なった。結晶学的パラメータを Table 4-6、主な結合長と結合角を Table 4-7、結晶構造の
ORTEP 図を Figure 4-5(left)に示した。得られた錯体はエクアトリアル平面にアザイドが一分
子配位した、単核の平面 4 配位型錯体であった。しかしながら、結晶構造において、配位
アザイド分子は隣接する錯体の平面下方より約 2.4Åの距離をもって弱く配位しており、一
次元鎖状構造を形成していた。ゆえに、結晶状態において錯体の三級アミン窒素-銅-アザイ
ド分子の角度は∠N(1)-Cu(1)-N(4) = 159.1(5)o であり若干の歪みが認められた。空配位座は
MeOH, MeCN, H2O 等の溶媒分子から、N3-イオンに置換することによりその配位結合距離が
約 0.08Å短くなっており、錯体はアニオン性配位子であるアザイド分子を溶媒分子より強
く捕捉していると考えられる。
4.3.4 [CuII(bpba)(MeOH)]2+錯体と Cl-イオンとの反応
[CuII(bpba)(MeOH)]2+錯体のメタノール溶液に 1 当量の KCl を添加し、UV-vis, ESR スペク
トル測定を行った。錯体濃度 0.2 mM における UV-vis スペクトルを Figure 4-4(right)に、ま
た UV-vis, ESR スペクトル的特徴を Table 4-5 にまとめた。
KCl の添加による溶液の色変化は見られず、この青色溶液は UV-vis スペクトルにおいて
λmax = 294 nm (ε = 2650 M-1cm-1), 687 nm (ε = 175 M-1cm-1)に特徴的な吸収帯を確認した。前者
69
は Cl-→CuII に相当する LMCT、後者は CuII 錯体に由来する d-d 遷移と帰属される。スペク
ト ル は 平 面 型 の 銅 (II) 錯 体 に 特 徴 的 で あ り 、 溶 液 中 に お い て 錯 体 は 平 面 4 配 位 型の
[CuII(bpba)(Cl-)]+として存在すると考えられる。ESR スペクトルを測定したところスペクト
ルは平面型の銅(II)錯体に特徴的な形状であり、g⊥=2.04, g//=2.22, |A//|=158 であった。|A//|値
より溶液状態において錯体は歪んだ平面構造であることが判明した。錯体は単結晶が得ら
れたため X 線による結晶構造解析を行なった。結晶学的パラメータを Table 4-6、主な結合
長と結合角を Table 4-7、結晶構造の ORTEP 図を Figure 4-5(right)に示した。得られた錯体は
空配位座にアザイドが一分子配位した、単核の平面 4 配位型錯体であった。結晶状態にお
いて錯体の三級アミン窒素-銅-塩化物イオンの角度は∠N(1)-Cu(1)-Cl(1) = 152.71o であり若
干の歪みが認められた。
4.3.5 [CuII(bpba)(H2O)]2+錯体と過酸化水素との反応
[CuII(bpba)(H2O)]2+錯体と過酸化水素との反応の UV-vis スペクトル変化を Figure 4-6 に、
ESR スペクトル変化を Figure 4-7 に示した。
[CuII(bpba)(H2O)]2+錯体のアセトン溶液に、Ar 雰囲気下-80 oC において 1.2 当量のトリエチ
ルアミンを添加すると溶液は青色から濃青色へと変化し、UV-vis スペクトル測定において
λmax = 345 nm (ε = 2290 M-1cm-1), 580 nm (ε = 76 M-1cm-1) , 793 nm (ε = 50 M-1cm-1)に特徴的な
極大吸収を観測した。そのスペクトル的特徴より[CuII2(bpba)2(µ-OH)2]2+錯体が生成したと考
えられ13、345 nm の吸収帯はヒドロキソイオンから CuII イオンへの LMCT と考えられ、580
nm, 793 nm に見られる吸収帯は銅 CuII イオンの d-d 遷移に由来すると帰属される。このとき
の ESR スペクトルは silent であり、複核錯体の生成が示唆された。別途調製した溶液から
は単結晶が得られたため、その X 線結晶構造解析を行った。
結晶学的パラメータを Table 4-8、
主な結合長と結合角を Table 4-9、結晶構造の ORTEP 図を Figure 4-8 に示した。
さらにこの[CuII2(bpba)2(µ-OH)2]2+錯体溶液に 5 当量の過酸化水素を添加すると溶液は緑色
へと変化し、UV-vis スペクトル測定においてλmax = 350 nm (ε = 3400 M-1cm-1), 564 nm (ε = 150
M-1cm-1), 790 nm (ε = 55 M-1cm-1)に特徴的な極大吸収を観測した(Figure 4-6)。350 nm の吸収
帯はハイドロパーオキソイオンから CuII イオンへの LMCT と考えられ、564 nm, 790 nm に
見られる吸収帯は CuII イオンの d-d 遷移に由来すると帰属される 3。ESR スペクトル測定を
行ったところ square planar 型構造を有する単核銅(Ⅱ)錯体に特徴的なシグナルを観測し、g// =
2.26, g⊥ = 2.06, |A//| = 175 G であった(Figure 4-7)。同条件において調製した錯体溶液の共鳴
ラマンスペクトル測定の結果を Figure 4-9 に示した。H216O2 を用いた測定において 834 cm-1
にパーオキソ種に由来すると考えられる O-O 伸縮振動を観測した。H218O2 を用いて同様の
測定を試みたが酸素分子に由来するピークの検出は困難であった。同条件において調製し
70
た錯体溶液の ESI-mass スペクトル測定の結果を Figure 4-10 に示した。m/z = 318.0 にペアレ
ントピークを観測し、これは[CuI(bpba)]+錯体の同位体パターンと一致した。[CuI(bpba)]+錯
体は銅(II)-ハイドロパーオキソ錯体の分解により生成したと考えられるが、その詳細は不明
で あ る 。 m/z = 351.0 に 観 測 さ れ る ピ ー ク は 単 核 銅 (II)- ハ イ ド ロ パ ー オ キ ソ 錯 体
([CuII(bpba)(OOH)+])の同位体パターンと一致した。ハイドロパーオキソ錯体は非常に不安定
であるためピークの相対強度は小さいが、これにより単核銅(II)-ハイドロパーオキソ錯体の
生成が確認された。これらスペクトル測定を総合し、単核銅(II)錯体([CuII(bpba)(OH2)]2+)と
過酸化水素との反応により、単核銅(II)-ハイドロパーオキソ錯体([CuII(bpba)(OOH)]+)が生成
したことが明らかとなった。
4.3.6
単核銅(II)-ハイドロパーオキソ錯体の熱力学的安定性
単核銅(II)-ハイドロパーオキソ錯体に特徴的な LMCT の減衰を追跡することにより、錯体
の分解速度を測定した。実験は、配位子に TPA を用いた 5 配位型の単核銅(II)-ハイドロパー
オキソ錯体([CuII(tpa)(OOH)]+)についても同様に行い、これらの比較により錯体構造がその
分解速度に及ぼす影響を検討した。この[CuII(tpa)(OOH)]+錯体はポリピリジルアミンを有す
る 5 配位三脚型錯体のうち、最も基本的な構造を有しており、比較対象として最適と考え
られる。
単核銅(II)-ハイドロパーオキソ錯体の合成と追跡は Stopped flow 法を用いて行った。Ar
雰囲気下にて調製した[CuII(bpba)(H2O)]2+, [CuII(tpa)(MeCN)]2+錯体のアセトン溶液(1 mM)に、
2 当量のトリエチルアミンを添加した反応溶液 A と、別途調製した過酸化水素濃度 100 mM
のアセトン溶液 B を温度 10 oC に制御した測定セル内で混合し、それぞれ 350 nm, 380 nm に
見られる特徴的な LMCT の時間変化を測定した(Figure 4-11 left)。各錯体の分解速度は一次
の速度式に従い、その擬一次速度定数(kobs)はそれぞれ 1.9 s-1 ならびに 8.3×10-2 s-1 であった
(Figure 4-11 right)。
4.3.7
単核銅(II)-ハイドロパーオキソ錯体と基質との反応
低温条件下において合成した[CuII(bpba)(OOH)]+錯体に過剰量の基質を添加することによ
り、外部基質に対する酸化反応を行った。[CuII(bpba)(OOH)]+錯体の合成は 4.3.5 項に従い、
アセトン溶媒(-80 oC)中で錯体に 1.2 当量のトリエチルアミンと 10 当量の過酸化水素を添加
することにより行った。[CuII(bpba)(OOH)]+錯体の合成後に 500 当量の基質をシリンジにて
添加し、反応の進行は 350 nm に見られる特徴的な LMCT の減衰により確認した。反応終了
後に過剰量のトリフェニルフォスフィン(PPh3)を添加し、徐々に室温に昇温することによっ
て未反応の過酸化水素を除去し、GC 測定により酸化生成物の定量を行った。各種基質に対
71
する反応結果を Table 4-10 に示した。
錯体はジメチルスルフィド、チオアニソール、エチルベンゼン、クメン、シクロヘキセ
ン等に対して反応性を示し、比較的酸化されにくい基質であるトルエン、シクロヘキサン
に対しては反応性を示さなかった。チオエーテル類の酸化については対応するスルフォキ
シドを酸化生成物として生成し、合成した銅(II)-ハイドロパーオキソ錯体に対し定量的な酸
化反応が進行した。エチルベンゼンの酸化反応においては 1-フェニルエチルアルコールが
30%、ベンズアルデヒドが 24%の収率で観測された。合成した単核銅(II)-ハイドロパーオキ
ソ錯体は酵素と同様に芳香環α位の水酸化反応に反応活性を有することが明らかとなった。
また、続く脱水素反応によりケトンが生成したと考えられる。同様に芳香環のα水素を有す
るクメンの酸化においてはクミルアルコールを収率 18%で観測した。クメンは反応点であ
るα水素周りの立体障害がエチルベンゼンより増しており、活性種との接触効率が低下する
ため、収率が減少したと考えられる。シクロヘキセンの酸化反応ではシクロヘキセノール
を収率 70%で、シクロヘキセノンを収率 22%で観測した。銅(II)-ハイドロパーオキソ錯体は
アリル位の水酸化反応、またそれに続くケトンの生成に対して反応性を有することが明ら
かとなった。シクロヘキセンの酸化反応では、もう一つの酸化生成物としてシクロヘキセ
ンオキシドが考えられる。この酸化生成物は活性種が M=Oδ+、すなわち原子状酸素の性質
を有する場合、オレフィンに対する求電子的な酸素分子挿入反応により生成する14。本反応
ではシクロヘキセンオキシドの生成は認められなかったことから、活性種がメタルオキソ
的な性質を有していないことが示唆された。これらの反応特性より、反応は銅(II)-ハイドロ
パーオキソ錯体の O-O 結合がホモリティックに開裂することによりラジカル種が生成し、
水素原子引き抜き反応とラジカル再結合反応により進行していることが明らかとなった。
4.3.8
触媒的酸化反応
4.3.7 項において、合成した銅(II)-ハイドロパーオキソ錯体([CuII(bpba)(OOH)]+)は各種基質
に対する反応性を有することが明らかとなった。そこで、過酸化水素を酸化剤とし、この
ような活性種を経由する触媒的な酸化反応を試みた。反応は Ar 雰囲気下にてアセトニトリ
ル溶液に、錯体:トリエチルアミン:基質 = 1:2:500 の割合で混合し、0 oC に冷却しな
がら錯体に対し 100 当量の過酸化水素を添加することにより行った。(詳細な反応条件は
4.2.5 項に示した。) GC 測定による酸化生成物の定量を行い、その結果を Table 4-11 に示し
た。
錯体はチオエーテルの酸化反応について高い触媒活性を示すことが明らかとなった。ジ
メチルスルフィドの酸化においては添加した過酸化水素に対し収率 70% (TON = 70)で反応
が進行し、チオアニソールについては収率 100% (TON = 100)で反応が進行した。このよう
72
な触媒反応において、どちらの錯体もスルホンの生成は確認されず、選択的な酸化反応が
進行した。シクロヘキセン、テトラリンに対するアリル位酸化においても触媒的な酸化反
応の進行を確認した。これらの反応では対応するアルコールが選択的に生成し、ケトンの
生成はごく僅かであった。また、シクロヘキセンの酸化反応では触媒条件下においてもシ
クロヘキセンオキシドの生成は認められず、ラジカルを中間体とした反応が進行している
と考えられる。シクロヘキセノールの収率は錯体の TON に換算して TON = 11.6、シクロヘ
キセノンの収率は TON = 1.6 であり、選択的なアルコールの生成を確認した。エチルベンゼ
ン、クメンのベンジル位の酸化における酸化活性は非常に低く、錯体はこれらの反応を効
率よく触媒することができなかった。エチルベンゼンの酸化ではベンジルアルコール(TON
= 2.1)がアセトフェノン(TON = 1.6)の収率を若干上回っていた。最も反応性の高かったチオ
アニソールを基質に用い反応条件の最適化を行った。10 当量の酸化剤を 5 分間隔で添加し
た場合における酸化反応生成物量の経時変化を Figure 4-12 (left)に示した。反応開始後 0~30
分において、反応は添加した過酸化水素に対し定量的に進行したが、そのあと錯体は徐々
に失活し、反応開始後 60 分では添加した過酸化水素に対し僅かに反応が進行するのみであ
った。この時、過酸化水素は錯体に対し 120 当量添加されているにもかかわらず TON は 92
であり、100 当量の過酸化水素を一度に添加した場合よりも低い収率を示した。酸化剤を
100 当量ずつ 15 分間隔で添加した場合の結果を Figure 4-12 (right)に示した。この場合も反
応は 0~30 分の間は添加した過酸化水素に対し定量的に進行し、さらに反応後 30~60 分に
かけて錯体は失活した。この時の過酸化水素の添加量は 400 当量であり、それに対する TON
は 300 (収率 75%)であった。さらに、基質を 2000 当量にスケールアップし 1000 当量の過酸
化水素を 1 分間かけて添加したところ TON が 470 に達した。
73
4.4
考察
4.4.1 4 配位型単核銅(II)-ハイドロパーオキソ錯体の合成
配位子 BPBA を用いて合成した単核銅(II)錯体は、溶液中において空配位座に溶媒分子が
配位した 4 配位平面型錯体を形成していた。ESR スペクトルによる|A//|の値を過去に報告さ
れている錯体群と比較することにより、錯体の平面構造はやや歪んでいることが判明して
いる
12
。Ar 雰囲気下において[CuII(bpba)(H2O)]2+錯体とトリエチルアミンを反応させたとこ
ろ複核の[CuII2(bpba)2(µ-OH)2]2+錯体を生成し、これに過酸化水素を添加することにより単核
の[CuII(bpba)(OOH)]+錯体が生成した(Scheme 4-1)。このような二核 bis-µ-hydroxo 錯体を経由
した単核銅(II)-ハイドロパーオキソ錯体の合成は、過去に Kodera らが報告している 3(a)。反
応後の UV-vis スペクトルにおいて観測された 350 nm (ε = 3400 M-1cm-1)の極大吸収は、ハイ
ドロパーオキソイオンから CuII イオンに対する LMCT と帰属されるが、これは過去に帰属
された銅(II)-ハイドロパーオキソ錯体の中でも短波長側に位置し、そのモル吸光係数はかな
り大きい値であった 3,4,5。また、d-d 吸収帯は 564 nm (ε =150 M-1cm-1), 790 nm (sh, ε = 55
M-1cm-1)に 2 つの極大吸収を観測した。ESR スペクトルは平面型の単核銅(II)錯体に特徴的な
スペクトルが観測され、また ESI-mass スペクトルにおいて[CuII(bpba)(OOH)]+に由来するピ
ークが観測されたことから、錯体は単核銅(II)-ハイドロパーオキソ錯体であると考えられる。
N3-イオンは分子の形状や配位性の軌道がハイドロパーオキソイオンに類似しており、銅(II)アザイド錯体はその安定性が高いことから、銅(II)-ハイドロパーオキソ錯体の構造推定に利
用されてきた 5。[CuII(bpba)(MeOH)]2+錯体と NaN3 の反応からは単核の[CuII(bpba)(N3-)]+錯体
が得られており、この結果からも単核銅(II)-ハイドロパーオキソ錯体の生成が示唆される。
4.4.2
[CuII(bpba)(OOH)]+錯体の基本的物性
過去に報告された各種銅(II)-ハイドロパーオキソ錯体の分光学的特徴を Table 4-12 にまと
めた。表中の錯体は大きく分けると、単核の銅(II)-ハイドロパーオキソ錯体と複核の銅(II)ハイドロパーオキソ錯体に分類される。複核銅(II)錯体による架橋ハイドロパーオキソ錯体
は古くは Karlin 等により報告され15、近年は Suzuki らにより架橋-OH, -OOH 錯体の結晶構造
が報告されている16。錯体は配位子の違いによりλmax = 340~395 nm の領域にハイドロパー
オキソイオンから銅(II)イオンに対する LMCT を観測し、共通の特徴としてε = 6000~12000
M
-1
cm-1 の比較的大きなモル吸光係数を有している。二核の銅(II)-ハイドロパーオキソ錯体
が単核に比べ大きいモル吸光係数を示す理由は、ハイドロパーオキソイオンをリジッドに
捕捉することよりそのπ*軌道から CuII(d)軌道への遷移確率が高くなるためと推測されてい
る17。これに対し、単核の銅(II)-ハイドロパーオキソ錯体のスペクトル的な特徴はλmax = 357
74
~ 380 nm の領域にε = 890 ~ 4300 M -1cm-1 の吸光係数で LMCT が観測されることである。こ
れらは全て 5 配位型の銅(II)-ハイドロパーオキソ錯体であるが、その構造上さらに細かく分
類すると、三方両錘構造である[CuII(H2bppa)(OOH)]+, [CuII(tpa)(OOH)]+錯体と、平面四角錐
構造である[CuII(N3S)(OOH)]+錯体に分けることができる。合成した[CuII(bpba)(OOH)]+錯体は
4 配位平面型の単核銅(II)-ハイドロパーオキソ錯体と考えられるが、そのスペクトル的特徴
は Kodera 等の報告した[CuII(N3S)(OOH)]+錯体に近いことがわかる 3-(a)。[CuII(N3S)(OOH)]+錯
体はハイドロパーオキソ酸素、2 つのピリジン窒素、1 つのアルキルアミン窒素で銅を中心
とした平面が構成されており、軸位よりチオエーテルの硫黄原子が配位していると考えら
れ 、 そ の 構 造 は 軸 配 位 子 を 除 け ば [CuII(bpba)(OOH)]+ 錯 体 に 類 似 し て い る 。 ま た 、
[CuII(N3S)(OOH)]+錯体もまた[CuII(bpba)(OOH)]+錯体と同様に dx2-y2 基底に由来した ESR スペ
クトルを示したことから、両錯体ともハイドロパーオキソイオンが銅(II)イオンの dx2-y2 軌道
に配位することも共通しており、よく似た分光学的特徴を示したと推定される。
アセトン溶媒中、10 oC における[CuII(bpba)(OOH)]+錯体の分解速度は一次の速度式に従い、
その擬一次速度定数(kobs)は 1.9 s-1 であり、過去に報告された 5 配位型の単核銅(II)-ハイドロ
パーオキソ錯体と比べ非常に大きい値であった。これらの 5 配位型錯体のうち最も基本的
な構造を有する[CuII(tpa)(OOH)]+錯体の分解速度を測定したところ、kobs = 8.3×10-2 s-1 であっ
た。錯体の分解はハイドロパーオキソイオンの O-O 間結合の開裂により進行すると考えら
れ、錯体の分解速度定数と共鳴ラマン測定による O-O 間結合の振動エネルギーには相関が
見られる。過去に報告された一連の単核銅(II)-ハイドロパーオキソ錯体とともに、錯体の分
解速度と O-O 間伸縮振動の相関関係を Figure 4-13 に示した。
グラフは良い直線関係を示し、
ハイドロパーオキソ錯体の分解速度が O-O 間結合エネルギーに比例していることが理解で
きる。すなわち、錯体の分解は配位ハイドロパーオキソイオンの O-O 間結合の開裂により
進行していると考えられる。従って、[CuII(bpba)(OOH)]+錯体の比較的早い分解速度は配位
しているハイドロパーオキソイオンの O-O 間結合が弱いことに起因する。錯体は 4 配位の
平面型構造でありハイドロパーオキソイオンは銅(II)イオンの dx2-y2 軌道と相互作用するこ
とにより配位結合を形成している。配位場理論より、この結合は 5 配位の三方両錘型錯体
よりも強いことが予想される。錯体は強い CuII-O 間結合を形成すると共にハイドロパーオ
キソイオンの O-O 間結合は弱くなり、錯体の分解が促進されたと考えられる18。
4.4.3 [CuII(bpba)(OOH)]+錯体の反応性
合成した単核銅(II)-ハイドロパーオキソ錯体の反応性を確認するために、様々な条件で基
質の酸化反応を行った。第一に、合成した活性種の厳密な反応特性を知るためには、非触
媒条件下で定量的な反応を行うことが望ましい。これは、触媒反応の進行に伴う系の複雑
75
化を防ぐことにより、より厳密な解析が可能となるためである。しかしながら、CuII-BPBA
錯体と過酸化水素による反応系は触媒的な酸化反応を進行させるが、この反応は低温条件
下で大きく制限することができる。そこで、低温条件下で少量の酸化剤を用いることによ
る非触媒的な酸化反応を行った。その結果、合成した[CuII(bpba)(OOH)]+錯体はジメチルス
ルフィド、チオアニソール、エチルベンゼン、クメン、シクロヘキセンに対して定量的な
反応性を示し、酸化されにくい基質であるトルエン、シクロヘキサンに対しては反応性を
示さなかった。また、アリル化合物の反応では対応するアルコールの生成が優先され、エ
ポキサイドの生成は確認できなかった。これにより反応活性種はラジカル的な性質を有し
ていることが予想され、このような化学種はハイドロパーオキソイオンの O-O 間結合がホ
モリティックに開裂することにより生成していると考えられる。反応系はこのようなラジ
カル種による水素原子引き抜き反応とラジカル再結合反応により、基質に対する酸素原子
の挿入が進行したと推定される。過去に合成された多くの 5 配位型の単核銅(II)-ハイドロパ
ーオキソ錯体は外部基質に対する反応性を示さず 4,5,19,20、また最も立体障害の少ない構造を
有する
[CuII(tpa)(OOH)]+錯体のジメチルスルフィドに対する反応性も、錯体に対し収率 5%
であった。4 配位型錯体と 5 配位型錯体間に見られる反応性の違いは銅(II)-ハイドロパーオ
キソ錯体自体の安定性と、錯体構造に起因する立体的な基質との接触し易さに依存してい
ると考えられる。このように合成した[CuII(bpba)(OOH)]+錯体は設計指針通り、外部基質に
対する反応性の向上に成功した。
また、各基質に対する反応性の差は、触媒条件下の酸化反応においてより大きく観測され
た。特にチオアニソールの酸化反応では、反応条件の改善により TON が 470 に達した。ま
た、過酸化水素を逐次的に添加して反応を行った場合、TON に関わらず錯体は 30 分程度で
失活が始まることが判明した。すなわち、時間の経過に伴い錯体が失活し、反応が停止し
ていると推定される。また、アリル位酸化やオレフィンの酸化に対する触媒活性は非常に
低く、基質により TON が 2 ~ 10 程度であった。これらの基質については非触媒条件下の反
応でも定量的な反応が進行しておらず、また継続的な過酸化水素の添加を行った場合も反
応の進行はごく僅かであった。すなわち、反応が円滑に進行しない基質は錯体自身の失活
を促進するため、錯体の触媒活性を低くしていると考えられる。比較のため[CuII(tpa)(OOH)]+
錯体を用いた触媒反応を行ったところ、同条件でのチオアニソールに対する反応性は TON =
1.6、シクロヘキセンに対する反応性は示さず、[CuII(bpba)(OOH)]+錯体に比べその反応性は
非常に低かった。このように、触媒反応系においても[CuII(bpba)(OOH)]+錯体は、5 配位型の
単核銅(II)-ハイドロパーオキソ錯体に比べ高い反応性を発現した。
76
4.5
結論
極低温下における[CuII(bpba)(H2O)]2+錯体と過酸化水素を反応により、単核の銅(II)-ハイド
ロパーオキソ錯体([CuII(bpba)(OOH)]+)が生成することを各種分光学的測定により確認した。
また、そのスペクトル的特徴により、[CuII(bpba)(OOH)]+錯体は平面型錯体であることが示
唆された。合成した[CuII(bpba)(OOH)]+錯体の熱力学的な安定性を、過去に報告されている 5
配位型の単核銅(II)-ハイドロパーオキソ錯体
3,4,5
と比較したところ、錯体の分解速度が著し
く増加していることが判明した。一連の単核銅(II)-ハイドロパーオキソ錯体の分解速度定数
と、錯体の O-O 間伸縮の振動エネルギーには相関が見られることから、これらハイドロパ
ーオキソ錯体の分解反応は、配位ハイドロパーオキソイオンの O-O 間結合の開裂により進
行していると考えられる。[CuII(bpba)(OOH)]+錯体は平面構造がもたらすハイドロパーオキ
ソイオンの強い配位により O-O 間結合が弱くなり、その結果、O-O 間結合の開裂による錯
体の分解が 5 配位型錯体に比べその分解が促進されたと考えられる。
低温条件下で、合成した[CuII(bpba)(OOH)]+錯体に外部基質を添加したところ、単核銅(II)ハイドロパーオキソ錯体に特徴的な LMCT の減衰に伴って基質の酸化が進行した。錯体は
チオエーテル類やオレフィンの酸化に対し高い反応性を示し、DβM に類似する芳香環α位
の水酸化についても反応性を示した。各種基質を用いた場合の酸化生成物の分布より、反
応はラジカル機構で進行していることが推定される。従って、配位ハイドロパーオキソイ
オンのホモリティックな開裂に伴い発生したラジカル種が基質を酸化していると考えられ
る。このようなプロセスは、過去に Klinman 等が提案した酵素の反応機構21と良い一致を示
している。さらに、[CuII(bpba)(OOH)]+錯体は反応条件の最適化により、外部基質に対して
触媒反応性を示し、チオエーテル、アリル化合物、芳香環α位の酸化反応活性を有すること
が明らかとなった。このようにモデル錯体を用いて合成した単核銅(II)-ハイドロパーオキソ
錯体が外部基質に対する触媒反応性を示した例はなく、大変興味深い。
今回、平面 4 配位型の単核銅(II)-ハイドロパーオキソ錯体を新たに合成し、過去に報告さ
れている 5 配位型の単核銅(II)-ハイドロパーオキソ錯体 3,4,5 と比較したところ、錯体の配位
構造が単核銅(II)-ハイドロパーオキソ錯体の安定性に非常に大きな影響を与えることが明
らかとなった。また、[CuII(bpba)(OOH)]+錯体は外部基質に対する反応性を示したことから、
錯体自身の不安定化や銅イオン周りの開けた反応空間がその反応性の向上につながったと
考えられる。
77
References
1
I. Bertini, A. Sigel and H. Sigel, Handbook on METALLOPROTEINS -Section 15-, Marcel
Dekker, Inc., New York, 2001.
2
(a) S. M. Miller and J. P. Klinman, Biochemistry, 1983, 22, 3091-3096. S. M. Miller and J. P.
Klinman, Biochemistry, 1985, 24, 2114-2127. (b) G. Tian, J. A. Berry and J. P. Klinman,
Biochemistry, 1994, 33, 226-234. (c) W. A. Francisco, D. J. Merkler, N. J. Blackburn and J. P.
Klinman, Biochemistry, 1998, 37, 8244-8252.
3
(a) M. Kodera, T. Kita, I. Miura, N. Nakayama, T. Kawata, K. Kano and S. Hirota, J. Am. Chem.
Soc., 2001, 123, 7715-7716. (b) P. Chen, K. Fujisawa and E. I. Solomon, J. Am. Chem. Soc.,
2000, 122, 10177-10193. (c) T. Osako, S. Nagatomo, Y. Tachi, T. Kitagawa and S. Itoh, Angew.
Chem. Int. Ed., 2002, 41, 4325-4328.
4
A. Wada, M. Harata, K. Hasegawa, K. Jitsukawa, H. Masuda, M. Mukai, T. Kitagawa and H.
Einaga, Angew. Chem. Int. Ed., 1998, 37, 798-800.
5
(a) S. Yamaguchi, S. Nagatomo, T. Kitagawa, Y. Funahashi, T. Ozawa, K. Jitsukawa and H.
Masuda, Inorg Chem, 2003, 42, 6969-6970. (b) S. Yamaguchi, A. Wada, S. Nagatomo, T.
Kitagawa, K. Jitsukawa and H. Masuda, Chem Lett, 2004, 33, 1556-1557. (c) S. Yamaguchi, A.
Kumagai, S. Nagatomo, T. Kitagawa, Y. Funahashi, T. Ozawa, K. Jitsukawa and H. Masuda, Bull.
Chem. Soc. Jpn., 2005, 78, 116-124.
6
(a) S. T. Prigge, A. S. Kolhekar, B. A. Eipper, R. E. Mains and L. M. Amzel, Science, 1997, 278,
1300-1305. (b) S. T. Prigge, B. A. Eipper, R. E. Mains and L. M. Amzel, Science, 2004, 304,
864-867.
7
P. Chen and E. Solomon, J. Am. Chem. Soc., 2004, 126, 4991-5000.
8
R. R. Jacobson, Z. Tyeklar, A. Farooq, K. D. Karlin, S. Liu and J. Zubieta, J. Am. Chem. Soc.,
1988, 110, 3690-3692.
9
International Tables for X-ray Crystallography, Ibers, J. A.; Hamilton, W. C. eds., Kynoch Press,
Birmingham, U. K., 1974, Vol. IV.
10 “Crystal Structure” analysis program (ver 3.7) Produced by Rigaku, 2005
11 A. W. Addison, T. N. Rao, J. Reedijik, J. Rijin, G. C. Vershoor, J. Chem. Soc. Dalton. Trans.,
1984, 7, 1349-1356.
12 (a) 化学総説, No.20, p179, 学芸出版センター, 東京, 1978. (b) 桜井
弘, ESR スペクトル
の実際, p132, 廣川書店, 1989.
13 S. Mahapatra, J. A. Halfen, E. C. Wilkinson, G. Pan, X. Wang, V. G. Young, Jr., C. J. Cramer, L.
Que, Jr. and W. B. Tolman, J. Am. Chem. Soc., 1996, 1118, 11555-11574.
14 (a) 松浦輝男, 「酸素酸化反応」, 丸善, 1977. (b) 中野稔, 浅田浩二, 大柳喜彦,「活性酸
素」, 共立出版, 1988. (c) 化学総説「活性酸素の化学」, 日本化学界編, 学術出版センタ
78
ー, 1990, 7
15 (a) K. D. Karlin, R. W. Cruse and Y. Gultneh, J. Chem. Soc., Chem Commun., 1987, 599-600.
(b) K. D. Karlin, P. Ghosh, R. W. Cruse, A. Farooq, Y. Gultneh, R. R. Jacobson, N. J. Blackburn,
R. W. Strange and J. Zubieta., J. Am. Chem. Soc., 1988, 110, 6769-6780. (c) M. Mahroof-Tahir,
N. N. Murthy, K. D. Karlin, N. J. Blackburn, S. N. Shaikh and J. Zubieta, Inorg Chem, 1992, 31,
3001-3003. (d) D. E. Root, M. Mahroof-Tahir, K. D. Karlin and E. I. Solomon, Inorg Chem,
1998, 37, 4838-4848. (e) L. Q. Hatcher and K. D. Karlin, J. Biol. Inorg. Chem., 2004, 9,
669-683.
16 K. Itoh, H. Hayashi, H. Furutachi, T. Matsumoto, S. Nagatomo, T. Tosha, S. Terada, S. Fujinami,
M. Suzuki and T. Kitagawa, J. Am. Chem. Soc., 2005, 127, 5212-5223.
17 R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239-2314.
18 S. Yamaguchi, Ph.D Dissertation of Nagoya Institute of Technology, 2004
19 M. Kodera, T. Kita, I. Miura, N. Nakayama, T. Kawata, K. Kano and S. Hirota, J. Am. Chem.
Soc., 2001, 123, 7715-7716.
20 P. Chen, K. Fujisawa and E. I. Solomon, J. Am. Chem. Soc., 2000, 122, 10177-10193.
21 (a) L. C. Stewart and J. P. Klinman, Annu. Rev. Biochem.,1988, 57, 551-592. (b) J. P. Klinman,
Chem. Rev., 1996, 96, 2541-2561.
79
O
HN
N
N
N
N
H
N
N
N
N
N
N
N
N
O
TPA
H2BPPA
BPBA
Chart 4-1 Ligands used this work.
Figure 4-1 ORTEP drawing of [CuII(H2bppa)(OOH)]+ with the atom labeling
scheme. The thermal ellipsoids are at the 50% probability level, and the
hydrogen atoms were omitted for clarity.
Table
4-1
Summary
of
the
spectroscopic
properties
of
[CuII(H2bppa)(OOH)]+
and
[CuII(tpa)(OOH)]+.
[CuII(H2bppa)(OOH)]+
[CuII(tpa)(OOH)]+
Electronic absorption (λmax/nm , ε/M-1cm-1)
380 (890); LMCT, 660 (150), 830 (250); d-d /MeCN
379 (1700); LMCT, 668 (170), 828 (200); d-d /MeCN
ESR
g⊥ = 2.21, g// = 2.00, |A⊥| = 75 G, |A//| = 109 G /MeOH
g⊥ = 2.29, g// = 2.01, |A⊥| = 87 G, |A//| = 99 G /MeCN
Resonance Raman (ν / cm-1)
873 cm-1(16O-16O), 826 cm-1(18O-18O) <∆ν 47 cm-1> /MeCN
847 cm-1(16O-16O), 792 cm-1(18O-18O) <∆ν 55 cm-1> /MeCN
ESI-mass (m/z)
584 [Cu(bnpa)(16O2H)]+, 588 [Cu(bnpa)(18O2H)]+ /MeCM
387 [Cu(bnpa)(16O2H)]+, 391 [Cu(bnpa)(18O2H)]+ /MeCM
80
Table 4-2 Crystallographic data and refinement parameters for [CuII(bpba)(MeOH)](ClO4)2,
[CuII(bpba)(MeCN)2](ClO4)2 and [CuII(bpba)(OH2)](ClO4)2.
Empirical formula
Formula mass
Crystal system
Space group
a [Å]
b [Å]
c [Å]
α [o]
β [o]
γ [o]
V [Å3]
Z
D calced . [g cm-3]
F (000)
µ [cm-1]
λ [Å]
T [oC]
No. of refls. Measured
No. of refls. Used [I > 2σ(I 0)]
No. of Variables
R 1 [a]/ R w [b]
GOF
[Cu(bpba)(MeOH)](ClO4)2
C68H100Cu4N12Cl8O36
535.82
monoclinic
Pc (No. 7)
13.7793(5)
19.3635(6)
17.3317(7)
106.171(2)
4441.4(3)
2
1.603
2200.00
12.76
0.71070
20.0
19166
17844
340
0.0590 / 0.1000
1.550
[Cu(bpba)(MeCN)2](ClO4)2
C20H27Cl2CuN5O8
599.91
triclinic
P -1 (#2)
9.749(11)
11.42(2)
12.900(15)
89.79(5)
111.000(9)
104.28(3)
1294(3)
2
1.540
618.00
11.037
0.71070
-100.0
10326
4693
351
0.0540 / 0.1641
1.002
[Cu(bpba)(OH2)](ClO4)2
C32H42Cl4Cu2N6O18
1067.62
triclinic
P -1 (#2)
9.530(12)
15.56(2)
16.27(2)
63.11(3)
89.92(6)
82.81(6)
2130.8(40)
2
1.664
1092.00
13.29
0.71070
-100.0
9205
2784
601
0.122/0.311
0.979
Table 4-3 Selected bond lengths ( Å ) and angles (o) of [CuII(bpba)(MeOH)](ClO4)2,
[CuII(bpba)(MeCN)2](ClO4)2 and [CuII(bpba)(OH2)](ClO4)2.
[Cu(bpba)(MeOH)](ClO4)2
Cu(1)-N(1)
2.064(2)
Cu(1)-N(2)
1.936(3)
1.964(3)
Cu(1)-N(3)
Cu(1)-O(1)
2.017(2)
[Cu(bpba)(MeCN)2](ClO4)2
Cu(1)-N(1)
2.085(3)
Cu(1)-N(2)
1.955(2)
Cu(1)-N(3)
1.961(2)
Cu(1)-N(4)
2.019(3)
Cu(1)-N(5)
2.277(3)
[Cu(bpba)(OH2)](ClO4)2
2.08(2)
Cu(1)-N(1)
1.90(2)
Cu(1)-N(2)
1.91(2)
Cu(1)-N(3)
2.03(2)
Cu(1)-O(1)
N(1)-Cu(1)-O(1)
N(2)-Cu(1)-N(3)
N(1)-Cu(1)-N(2)
N(1)-Cu(1)-N(3)
N(2)-Cu(1)-O(1)
N(3)-Cu(1)-O(1)
N(1)-Cu(1)-N(2)
N(1)-Cu(1)-N(3)
N(1)-Cu(1)-N(4)
N(1)-Cu(1)-N(5)
N(2)-Cu(1)-N(3)
N(2)-Cu(1)-N(4)
N(2)-Cu(1)-N(5)
N(3)-Cu(1)-N(4)
N(3)-Cu(1)-N(5)
N(4)-Cu(1)-N(5)
N(1)-Cu(1)-O(1)
N(2)-Cu(1)-N(3)
N(1)-Cu(1)-N(2)
N(1)-Cu(1)-N(3)
N(2)-Cu(1)-O(1)
N(3)-Cu(1)-O(1)
179.68(10)
167.8(1)
84.01(10)
85.1(1)
96.0(1)
94.9(1)
81
84.38(11)
84.24(11)
160.95(13)
112.92(14)
164.30(12)
93.88(13)
97.41(11)
93.34(12)
96.98(12)
86.13(15)
160.6(8)
166.6(7)
82.6(8)
84.7(7)
95.0(8)
95.6(8)
Figure 4-2 ORTEP drawings of [CuII(bpba)(X)](ClO4)2 (X = MeOH, MeCN, H2O) with the
atom labeling scheme. The thermal ellipsoids are at the 50% probability level, and the
hydrogen atoms were omitted for clarity.
(top) [CuII(bpba)(MeOH)]2+, (middle) [CuII(bpba)(MeCN)2]2+, (bottom) [CuII(bpba)(OH2)]2+
82
0.5
Acetone
MeCN
MeOH
Absorbance
0.4
0.3
0.2
0.1
0
300
400
500
600
700
800
900
Wavelength [nm]
Figure 4-3 UV-vis spectra of [CuII(bpba)(X)]2+ (1 mM) in solutions.
X = MeOH
MeCN
MeOH solution・・・solid line
MeCN solution・・・dotted line
H2O Acetone solution・・・bold dotted line
Table 4-4 Spectroscopic features of [CuII(bpba)(X)]2+ in solutions. (X = MeOH, H2O).
complex / solvent
II
d-d (nm) / ε (M-1cm-1)
g⊥
g//
|A//|
650 (117)
640 (128)
2.27
2.25
2.07
2.08
154
153
2+
[Cu (bpba)(MeOH)] / MeOH
[CuII(bpba)(H2O)]2+ / Acetone
Table 4-5 Spectroscopic features of [CuII(bpba)(N3-)]+ and [CuII(bpba)(Cl-)]+ in MeOH solution.
complex
[Cu (bpba)(N3-)]+
II
- +
[Cu (bpba)(Cl )]
II
-1
-1
-1
-1
LMCT/ nm (ε/ M cm ) d-d/nm (ε/ M cm )
396 (2870)
657 (360)
294 (2650)
687 (175)
83
g⊥
2.05
2.04
g//
2.22
2.22
|A//|
177
158
0.8
0.7
0.7
0.6
0.6
0.5
0.5
Absorbance
Absorbance
0.8
0.4
0.3
0.4
0.3
0.2
0.2
0.1
0.1
0
300
400
500
600
700
800
900
Wavelength [nm]
0
200
300
400
500
600
700
800
900
Wavelength [nm]
Figure 4-4 UV-vis spectra of [CuII(bpba)(N3-)]+ (0.2 mM) (left) and [CuII(bpba)(Cl-)]+ ( 0.2 mM)
(right) in MeOH solution.
Table 4-6 Crystallographic data and refinement parameters for [CuII(bpba)(N3-)](ClO4) and
[CuII(bpba)(Cl-)](ClO4).
Empirical formula
Formula mass
Crystal system
Space group
a [Å]
b [Å]
c [Å]
α [o]
β [o]
γ [o]
V [Å3]
Z
D calced . [g cm-3]
F (000)
µ [cm-1]
λ [Å]
T [oC]
No. of refls. Measured
No. of refls. Used [I > 2σ(I 0)]
No. of Variables
R 1 [a]/ R w [b]
GOF
[Cu(bpba)(N3)]ClO4
C32H42Cl2Cu2N12O8
920.76
triclinic
P -1 (#2)
9.778(2)
15.769(4)
16.512(4)
82.245(11)
74.632(9)
76.207(10)
2377.2(10)
2
1.286
948.00
10.60
0.71070
-100.0
19093
4055
547
0.069 / 0.195
1.003
84
[Cu(bpba)(Cl)]ClO4
C16H21Cl2CuN3O4
453.81
monoclinic
C 2/c (#15)
10.668(12)
14.466(14)
26.01(3)
96.99(3)
3984(8)
8
1.513
1864.00
13.897
0.71070
-100.0
15416
3727
256
0.0857 / 0.2566
1.000
Table
II
4-7
Selected
bond
lengths
and
angles
of
[CuII(bpba)(N3-)](ClO4)2
and
-
[Cu (bpba)(Cl )](ClO4)2.
[Cu(bpba)(Cl)]ClO4
[Cu(bpba)(N3)]ClO4
Bond lengths (Å)
Cu(1)-N(1)
2.071(8) Cu(1)-N(1)
2.061(3)
Cu(1)-N(2)
1.977(8) Cu(1)-N(2)
1.965(4)
Cu(1)-N(3)
1.960(8) Cu(1)-N(3)
1.966(3)
Cu(1)-N(4)
1.94(1)
Cu(1)-Cl(1)
2.2119(14)
N(1)-Cu(1)-N(2)
N(1)-Cu(1)-N(3)
N(1)-Cu(1)-N(4)
N(2)-Cu(1)-N(3)
N(2)-Cu(1)-N(4)
N(3)-Cu(1)-N(4)
Bond angles ( o )
85.0(3)
N(1)-Cu(1)-N(2)
84.6(3)
N(1)-Cu(1)-N(3)
159.1(5) N(1)-Cu(1)-Cl(1)
164.9(4) N(2)-Cu(1)-N(3)
101.9(4) N(2)-Cu(1)-Cl(1)
91.7(4)
N(3)-Cu(1)-Cl(1)
84.32(16)
84.99(15)
152.71(13)
159.20(18)
98.82(13)
98.82(13)
Figure 4-5 ORTEP drawing of [CuII(bpba)(X)]ClO4 (X = N3-, Cl-) with the atom labeling scheme.
The thermal ellipsoids are at the 50% probability level, and the hydrogen atoms were omitted for
clarity.
(left) [CuII(bpba)(N3-)]ClO4,
(right) [CuII(bpba)(Cl-)]ClO4
85
Absorbance
1.6
1.2
0.8
0.4
0
300 400 500 600 700 800 900 1000 1100
Wavelength [nm]
Figure 4-6 UV-vis spectral change of the reaction of [CuII(bpba)(OH2)]2+ with Et3N
and H2O2 in acetone at -80 oC.
(solid line) [CuII(bpba)(OH2)]2+ 0.5 mM, (dotted line) addition of Et3N 1.2eq into
the complex solution, (bold line) addition of H2O2 5eq into the complex solution
Figure 4-7 ESR spectral change of the reaction of [CuII(bpba)(H2O)]2+ with Et3N
and H2O2 in acetone.
(top) [CuII(bpba)(H2O)]2+ (1 mM) with Et3N 1.2eq
(bottom) addition of H2O2 10eq into the complex solution
86
Table 4-8 Crystallographic data and refinement parameters for [CuII2(bpba)2(µ-OH)2](ClO4)2.
[Cu2(bpba)2(µ-OH)2](ClO4)2
C18H26CuN8ClO5
533.45
triclinic
P 1 (No. 2)
10.2639(2)
11.0910(5)
11.3520(1)
70.79(2)
75.30(2)
63.59(1)
1084.1(2)
2
1.634
522.00
11.81
0.71070
-100.0
340
4262
262
0.083 / 0.172
1.870
Empirical formula
Formula mass
Crystal system
Space group
a [Å]
b [Å]
c [Å]
o
α[ ]
o
β[]
γ [o]
3
V [Å ]
Z
-3
D calced . [g cm ]
F (000)
µ [cm-1]
λ [Å]
o
T [ C]
No. of refls. Measured
No. of refls. Used [I > 3σ(I 0)]
No. of Variables
[a]
[b]
R1 / Rw
GOF
Table 4-9 Selected bond lengths and angles of [CuII2(bpba)2(µ-OH)2](ClO4)2.
Cu(1)-O(1)
Cu(1)-N(1)
Cu(1)-N(3)
[Cu2(bpba)2(µ-OH)2](ClO4)2
Bond lengths (Å)
1.933(2)
Cu(1)-O(1)*
2.354(2)
Cu(1)-N(2)
1.993(2)
O(1)-Cu(1)-N(1)
O(1)-Cu(1)-N(2)
O(1)-Cu(1)-N(3)
N(1)-Cu(1)-N(2)
N(2)-Cu(1)-N(3)
Bond angles ( o )
117.67 (9)
O(1)*-Cu(1)-N(1)
164.6(1)
O(1)*-Cu(1)-N(2)
94.73(9)
O(1)*-Cu(1)-N(3)
77.79(9)
N(1)-Cu(1)-N(3)
93.2(1)
O(1)-Cu(1)-O(1)*
1.929(2)
2.009(2)
109.12(9)
92.92(9)
169.76(9)
80.20(9)
77.33(9)
Figure 4-8 ORTEP drawing of [CuII2(bpba)2(µ-OH)2](ClO4)2 with the atom labeling scheme. The
thermal ellipsoids are at the 50% probability level, and the hydrogen atoms were omitted for clarity
87
A : [Cu(bpba)(H2O)](ClO4)2 / acetone
B : + Et3N + H216O2
λex = 406.7 nm
834
B
A
950
9 00
8 50
R am an Shift / cm
8 00
7 50
-1
Figure 4-9 Resonance Raman spectra for the reaction product of [CuII(bpba)(OH2)]2+ with Et3N and
H216O2 in acetone at -80 oC.
Simulation of [Cu(bpba)(OOH)]+
318.0
351.0
Figure 4-10 ESI-mass spectrum for the reaction product of [CuII(bpba)(OH2)]2+ with Et3N and H2O2
in acetone at -80 oC.
88
0
1.6
-0.2
1.4
-0.4
ln((At-Ainf)/(A0-Ainf))
Absorbance (350 nm)
1.8
1.2
1
0.8
0.6
-0.6
-0.8
-1
-1.2
0.4
0
0.5
1
1.5
2
2.5
3
3.5
-1.4
4
0
0.1
0.2
0.3
time [s]
0.6
0.7
0.8
(Plots were exhibited 40 ms interval)
1
0
0.8
-0.5
ln((At-Ainf)/(A0-Ainf))
Absorbance (380 nm)
0.5
time [s]
(Plots were exhibited 40 ms interval)
0.6
0.4
0.2
0
0.4
-1
-1.5
-2
0
5
10
15
20
25
-2.5
30
0
5
10
15
20
25
30
time [s]
time [s]
(Plots were exhibited 0.3 s interval)
(Plots were exhibited 0.3 s interval)
Figure 4-11 Representative spectral changes for decomposition process of copper(II)-hydroperoxo
species in acetone at 10 oC. <left column> Time courses for the characteristic absorption change of
BPBA (top) and TPA (bottom). <right column> First-order kinetics plots for BPBA (top) and TPA
(bottom).
89
Table 4-10 Summary of the oxidation reaction by the [CuII(bpba)(OOH)]+ under low temperature
experiment (-80 oC). Yield based on the generated copper(II)-hydroperoxo species.
Substrates
Products (GC yield%)
O
S
S
(100%)
S
(50eq)
O
S
(100%)
(50eq)
OH
(200eq)
O
(30%)
(24%)
OH
(200eq)
(18%)
(200eq)
NR
O
OH
(200eq)
(70%)
(200eq)
(22%)
NR
Table 4-11 Summary of the catalytic oxidation by the CuII-BPBA complex with hydrogen peroxide
system. TON exhibits a “turn over number”.
Substrates
Products (TON)
(70)
O
S
S
O
S
S
(100)
OH
O
(2.1)
(1.6)
OH
(2.4)
O
OH
(11.6)
O
OH
(8.5)
90
(1.6)
(2.6)
120
400
350
100
300
250
TON
TON
80
60
200
150
40
100
20
50
0
0
0
10
20
30
40
50
0
60
10
20
30
40
50
60
time [min]
time [min]
Figure 4-12 Correlation between catalytic reactivity and reaction time in the case of thioanisole
oxidation. (left) Hydrogen peroxide (10 eq) was added every 5 min. (right) Hydrogen peroxide (100
eq) was added every 15 min.
N
N
H
O
Et3N
Cu
LCu
OH2
N
CuL
N
H2O2
N
Cu
O
H
O
N
Scheme 4-1 Reaction scheme of [CuII(bpba)(H2O)]2+ with Et3N and H2O2.
Table 4-12 Spectroscopic data for mononuclear and dinuclear copper(II)-hydroperoxo complexes.
complexes
[Cu2(L3)2(OOH)(OH)]
2+
UV-vis λmax/nm (ε/M-1cm-1)
356 (6300), 582 (240), 664 (sh~190)
16-18
ν(O-O) (
16-18
∆), ν(Cu-O) (
868 (45), 572 (16)
[Cu2(L1)2(OOH)(OH)]2+
341(~7000), 581 (~170), 770 (sh~80)
883 (50), 562 (23)
[Cu2(UN-O)(OOH)]2+
395 (7000), 645 (660)
892 (55), 506 (15), 332 (10)
[Cu2(L)(OOH)]3+
342 (12000), 444 (1200), 610 (800)
[Cu(tpa)(OOH)]+
[Cu(H2bppa)(OOH)]+
379 (1700), 668 (170), 828 (200)
380 (890), 660 (150), 830 (250)
847 (55)
856 (46)
[Cu(N3S)(OOH)]+
357 (4300), 600 (140)
881 (49)
[Cu(HB(3-tBu-5-iPrpz)3(OOH)]+
600 (1540), 830 (~300)
843 (44), 624 (17)
L1 = bis(1-methyl-2-phenyl-4-imidazalylmethyl)benzylamine
L3 = tris(1-methyl-2-phenyl-4-imidazalylmethyl)amine
L = α,α'-bis{bis[2-(1'-methyl-2'-benzylimidazolyl)ethyl]-amino}-m -xylene
N3S = 2-bis(6-methyl-2-pyridylmethyl)amino-1-(phenylthio)ethane
HB(3-tBu-5-iPrpz)3 = hydrotris-(3-tert-butyl-5-isopropylpyrazolyl)borate anion
91
-1
∆) ν/cm
ref
OH
Figure 4-13 Correlation between log kobs and v(O-O) vibration from rRaman.
92
第5章
異なった平面性を有する単核銅(II)-ハイドロパーオキソ錯体
の合成と反応性の検討
- 4 配位型錯体間での構造と反応性の相関 5.1
序論
単核銅(II)-ハイドロパーオキソ種は、活性中心に銅イオンを含有する酸化酵素 DβM, PHM
の反応中間体として推定される化学種の一つである1。過去に合成されたモデル錯体は一般
的に不安定である単核銅(II)-ハイドロパーオキソ種を安定化させるために、銅イオン近傍に
疎水場や水素結合を導入しており、その安定性ゆえに基質との反応性を示さなかった2。ま
た、合成された単核銅(II)-パーオキソ錯体のほとんどが 5 配位型の錯体であるのに対し、
PHM の活性中心の構造は CuB イオン周りが 4 配位型の配位構造であることが明らかとなっ
ている3。また、その反応サイクルにおいて酵素の CuB サイトは 4 配位から 5 配位の配位構
造をフレキシブルに変化させていることが指摘されている4。そこで前章においては、新た
に合成した平面 4 配位型の単核銅(II)錯体と過酸化水素の反応による単核銅(II)-ハイドロパ
ーオキソ錯体の合成を行い、過去に報告されている 5 配位型錯体との比較を行なった。各
種分光学的測定より、[CuII(bpba)(OOH)]+錯体は歪んだ平面構造を有する単核銅(II)-ハイドロ
パーオキソ錯体であることが明らかとなり、その分解速度は過去に報告されている 5 配位
型錯体より増加していた。興味深いことに、合成した銅(II)-ハイドロパーオキソ錯体は外部
基質に対する反応性を示し、錯体の配位構造の違いが活性種の性質に大きな影響を与える
ことが明らかとなった。
本章では、BPBA と基本的な構造は同じだが、異なるアルキルアミン置換基を有する 3 座
型配位子を用いた 4 配位型の単核銅(II)錯体を合成した(Chart 5-1)。これら一連の 4 配位型錯
体はアルキル置換基の立体障害の大きさの違いにより、錯体の平面構造に系統的な差異が
生じることが予想される。先に述べたように、酵素はその反応機構の中で CuB サイトの構
造を変化させていることが指摘されており、錯体の配位構造の僅かな変化が単核銅(II)-ハイ
ドロパーオキソ錯体に与える影響をモデル化学的に検討することは非常に興味深い。そこ
で本章では、4 配位型錯体間の構造変化がその物性・反応性に及ぼす影響を検討することを
目的とした。このような検討は酵素の反応機構解明につながるのみならず、活性種を厳密
に制御し得る高機能触媒を開発する上で基礎的な知見を与えるものと考えられる。
93
5.2
実験
5.2.1
配位子合成
用いた三座型配位子の構造を Chart 5-1 に示した。
5.2.1.1
N,N’-Bis(2-pyridylmethyl)-iso-propylamine (BPIPA) の合成
1,4-ジオキサン 100ml に iso-propylamine 1.18 g (2.00×10-2 mol), (2-chloromethyl) pyridine
hydrochloride 8.2 g (5.00×10-2 mol)、蒸留水 20 ml に溶解させた KOH 5.62 g (0.1mol) を加え
pH 8~9 の条件下にて 80oC に加温、TLC (展開溶媒;クロロホルム:メタノール=7:1) で反
応の進行を確認しながら約 24 時間攪拌した。反応の進行に伴い KCl が析出し、反応溶液は
褐色を呈した。濾過により KCl を除去したのち、減圧濃縮により暗褐色の油状混合物を得
た。これを酢酸エチルに溶解させ 0.1N HCl aq で中和したのち有機相を分取、飽和食塩水、
無水 MgSO4 を用いて脱水処理をしたのち、減圧濃縮してシリカゲルカラム (溶離液;クロ
ロホルム:メタノール= 100:0~2) により単離した。得られた黄色油状物はヘキサン、エーテ
ル等を用いて結晶化を行い、淡褐色の針状結晶 2.84 g を得た。Yield 59%
1
H NMR (CDCl3, 300MHz ppm from TMS) ; 1.12(d, J (H-H)= 6.6 Hz, 6H, -CH3), 2.97(sep, J
(H-H)= 6.6 Hz, 1H, -CH-), 3.82(s, 4H, -CH2-), 7.11(t, J (H-H)= 6.0 Hz, 2H, Py-Hb),7.56~7.66(m,
4H, Py-Hc, Hd), 8.48(d, J (H-H)= 4.8 Hz, 2H, Py-Ha)
5.2.1.2
N,N’-Bis(2-pyridylmethyl)-ethylamine (BPEA) の合成
1,4- ジ オキ サ ン 150ml ethylamine(33 wt%aq) 2.73 g (2.00 ×10-2 mol), 2-(chloromethyl)
pyridine hydrochloride 7.2 g (4.40×10-2 mol)、蒸留水 20 ml に溶解させた KOH 4.94 g (8.8×10-2
mol) を加え pH 8~9 の条件下にて 60oC に加温、TLC (展開溶媒;クロロホルム:メタノール
=7:1) で反応の進行を確認しながら約 24 時間攪拌した。反応の進行に伴い KCl が析出し、
反応溶液は褐色を呈した。濾過により KCl を除去した後、減圧濃縮をして暗褐色の油状混
合物を得た。これを酢酸エチルに溶解させ 0.1N HCl aq で中和したのち抽出、飽和食塩水、
無水 MgSO4 を用いて脱水処理をした。これを濾過し、濾液を減圧濃縮してシリカゲルカラ
ム (溶離液;クロロホルム:メタノール= 100:0~2) により単離し褐色油状物 1.43 g を得た。
Yield 32%
1
H NMR (CDCl3, 300MHz ppm from TMS) ; 1.11(t, J (H-H)= 7.2 Hz, 3H, -CH3), 2.64(q, J (H-H)=
7.2 Hz, 2H, -CH2-), 3.82(s, 4H, -CH2-Py), 7.14(t, J (H-H)= 5.9 Hz, 2H, Py-Hb), 7.55(d, J (H-H)= 7.8
Hz, 2H, Py-Hd), 7.65(t, J (H-H)= 7.5 Hz, 2H, Py-Hc), 8.39(d, J (H-H)= 5.1 Hz, 2H, Py-Ha)
94
5.2.2
錯体合成
5.2.2.1 [CuII(bpipa)(MeOH)](ClO4)2 錯体の合成
配位子 BPIPA 14.48 mg (6×10-2 mmol)と Cu(ClO4)2·6H2O 22.23 mg (6×10-2 mmol)をメタノ
ール 0.15 ml にそれぞれ溶解させ、この溶液を混合したところ濃青色の溶液を得た。ジエチ
ルエーテル拡散により結晶化を行い、青色針状の結晶 23.1 mg を得た。
Yield: 72 %. Anal. Calcd. for [CuII(bpipa)(H2O)](ClO4)2 (C15H19Cl2CuN3O8⋅H2O): C, 34.53; H, 4.06;
N, 8.05. Found: C, 34.43; H, 4.16; N, 7.98.
5.2.2.2 [CuII(bpipa)(MeCN)](ClO4)2 錯体の合成
5.2.2.1 において得られた[CuII(bpipa)(MeOH)](ClO4)2 錯体をアセトニトリル溶媒に溶かし、
ジエチルエーテル拡散により再結晶させることにより濃青色の板状結晶を得た。
Yield: 97%. Anal. Calcd. for [CuII(bpipa)(H2O)](ClO4)2 (C15H19Cl2CuN3O8⋅H2O): C, 34.53; H, 4.06;
N, 8.05. Found: C, 34.40; H, 3.96; N, 8.16.
5.2.2.3 [CuII(bpipa)(H2O)](ClO4)2 錯体の合成
5.2.2.1 にて得られた[CuII(bpipa)(MeOH)](ClO4)2 錯体をアセトン溶媒に溶かし、ジエチルエ
ーテル拡散により再結晶させることにより濃青色の板状結晶を得た。
Yield: 98%. Anal. Calcd. for [CuII(bpipa)(H2O)](ClO4)2 (C15H19Cl2CuN3O8⋅H2O): C, 34.53; H, 4.06;
N, 8.05. Found: C, 34.69; H, 4.14; N, 7.95.
5.2.2.4 [CuII(bpea)(MeOH)](ClO4)2 錯体の合成
配位子 BPEA 13.64 mg (6×10-2 mmol)と Cu(ClO4)2·6H2O 22.23 mg (6×10-2 mmol)をメタノー
ル 0.15 ml にそれぞれ溶解させ、この溶液を混合したところ濃青色の溶液を得た。ジエチル
エーテル拡散により結晶化を行い、青色針状の結晶 16.3 mg を得た。
Yield: 52 %. Anal. Calcd. for [CuII(bpea)(H2O)](ClO4)2 (C14H17Cl2CuN3O8⋅H2O): C, 33.12; H, 3.77;
N, 8.28. Found: C, 32.82; H, 3.79; N, 8.00.
5.2.2.5 [CuII(bpea)(MeCN)](ClO4)2 錯体の合成
配位子 BPEA 13.64 mg (6×10-2 mmol)と Cu(ClO4)2·6H2O 22.23 mg (6×10-2 mmol)をアセトニ
トリル 0.15 ml にそれぞれ溶解させ、この溶液を混合したところ濃青色の溶液を得た。ジエ
チルエーテル拡散により結晶化を行い、紫色板状の結晶 17.5 mg を得た。
95
Yield: 55 %. Anal. Calcd. for [CuII(bpea)(H2O)](ClO4)2 (C14H17Cl2CuN3O8⋅H2O): C, 33.12; H, 3.77;
N, 8.28. Found: C, 33.25; H, 3.77; N, 8.21.
5.2.2.6 [CuII(bpea)(H2O)](ClO4)2 錯体の合成
配位子 BPEA 13.64 mg (6×10-2 mmol)と Cu(ClO4)2·6H2O 22.23 mg (6×10-2 mmol)アセトン
0.15 ml にそれぞれ溶解させ、この溶液を混合したところ濃青色の溶液を得た。ジエチルエ
ーテル拡散により結晶化を行い、青色板状の結晶 14.9 mg を得た。
Yield: 49 %. Anal. Calcd for [CuII(bpea)(H2O)](ClO4)2 (C14H17Cl2CuN3O8⋅H2O): C, 33.12; H, 3.77;
N, 8.28. Found: C, 33.37; H, 3.85; N, 8.12.
5.2.2.7 [CuII(bpipa)(N3)]ClO4 錯体の合成
単離した[CuII(bpipa)(MeOH)](ClO4)2 錯体 16.1 mg (3×10-2 mmol)をメタノール溶媒 10 ml に
溶かし、これに NaN3 1.95 mg (3×10-2 mmol)を加え超音波をかけながら良く攪拌した。配位
子置換反応の進行に伴い NaN3 は徐々に溶解し、溶液は青色から濃緑色に変化した。減圧濃
縮により得た濃緑色油状物を再度少量のメタノールに溶かしエーテル拡散を行うことによ
り青色板状の結晶を得たので X 線結晶構造解析による構造同定を行った。
5.2.2.8 [CuII(bpea)(N3)]ClO4 錯体の合成
単離した[CuII(bpea)(MeOH)](ClO4)2 錯体 15.7 mg (3×10-2 mmol)をメタノール溶媒 10 ml に溶
かし、これに NaN3 1.95 mg (3×10-2 mmol)を加え超音波をかけながら良く攪拌した。配位子
置換反応の進行に伴い NaN3 は徐々に溶解し、溶液は青色から濃緑色に変化した。減圧濃縮
により得た濃緑色油状物を再度少量のメタノールに溶かしエーテル拡散を行うことにより
青色板状の結晶を得たので X 線結晶構造解析による構造同定を行った。
5.2.2.9 [CuII(bpipa)(Cl)](ClO4)2 錯体の合成
単離した[CuII(bpipa)(MeOH)](ClO4)2 錯体 16.1 mg (3×10-2 mmol)をメタノール溶媒 10 ml に
溶かし、これに KCl 2.24 mg (3×10-2 mmol)を加え良く攪拌した。配位子置換反応の進行に
伴い、溶液の青色はやや薄く変化した。減圧濃縮により得た青色油状物を再度少量のメタ
ノールに溶かしエーテル拡散を行うことにより青色板状の結晶を得たので X 線結晶構造解
析による構造同定を行った。
5.2.2.10 [CuII(bpea)(Cl)](ClO4)2 錯体の合成
単離した[CuII(bpea)(MeOH)](ClO4)2 錯体 15.7 mg (3×10-2 mmol)をメタノール溶媒 10 ml に
溶かし、これに KCl 2.24 mg (3×10-2 mmol)を加え良く攪拌した。配位子置換反応の進行に
96
伴い、溶液の青色はやや薄く変化した。減圧濃縮により得た青色油状物を再度少量のメタ
ノールに溶かしエーテル拡散を行うことにより青色板状の結晶を得たので X 線結晶構造解
析による構造同定を行った。
5.2.3
銅(II)錯体と過酸化水素との反応
Ar 雰囲気下において調製した銅(II)錯体のアセトンもしくはアセトニトリル溶液 0.5 mM
に 2 当量のトリエチルアミンを加えこれを A 液とした。さらに Ar 雰囲気下にて過酸化水素
濃度 5.0 mM に調製したアセトンもしくはアセトニトリル溶液を B 液とした。これらの溶液
を 10 oC ~ -80 oC の範囲内で厳密に温度制御された Stopped flow 分光装置内で混合し、その
スペクトル変化を測定した。
5.2.4
触媒反応
銅(II)錯体 4 µmol、基質 2 mmol、GC 測定用の内部標準物質をアセトンもしくはアセトニ
トリル溶媒 2 ml に溶解させ反応容器に封入し、Ar 置換を行なった。これを ice bath 中 0 oC
に冷却後、トリエチルアミン 8 µmol、過酸化水素水 0.4 mmol をシリンジにて添加して 15
分間攪拌することにより反応を行なった。反応終了後、反応溶液約 50 µl を取り出し、過剰
量の PPh3 を添加したのち室温まで昇温させ、ガスクロマトグラフ測定により反応生成物の
定量を行なった。
5.2.5
5.2.5.1
測定装置
紫外可視吸収(UV-vis)スペクトル
測定装置は、日本分光製 Ubest-V570 紫外可視吸収分光光度計を使用し、測定セルは光路
長 1 cm の石英セルを使用した。サンプルは濃度を 0.25 ~ 1 mM の範囲で調製した錯体溶液
を用い、波長領域 900 ~ 300 nm の範囲において測定を行った。また、極低温での測定につ
いては UNISOKU の低温測定装置を分光器に取り付け、温度制御を行った。
5.2.5.2
電子スピン共鳴(ESR)スペクトル
測定装置は、JEOL JES-RE 1X ESR spectrometer を使用した。サンプルは濃度 1mM に調製
した錯体溶液を市販の ESR サンプルチューブに充填し、液体窒素を満たした測定用デュワ
ーに挿入し凍結させたのち、デュワーごと共振器に取り付けて測定を行った。測定条件を
以下に示した。Field, 3200±1000 G; Power, 1 mW; Sweep Time, 4 min; Modulation, 0.63 GHz;
Time Constant, 0.03 sec.
97
5.2.5.3 有機微量元素分析
測定装置は、Perkin Elmer 社製 2400II CHNS/O を使用した。試料測定前にガスブランク測
定を行った後、スズカプセルに封入した試料 1.5 ~ 2.0 mg を 2 回測定し、それを元素分析用
アセトアニリド標準試料による補正を行うことで C,H,N の各元素含有量(%)を求めた。
5.2.5.4
核磁気共鳴(1H-NMR)スペクトル
測定装置は、Varian Gemini 200 XL-300 型フーリエ変換核磁気共鳴装置を使用した。ケミ
カルシフトの基準物質として、テトラメチルシラン(TMS)を用いた。内径 5mmφ のサンプル
チューブ内に濃度を約 10 mM に調製した試料溶液について、積算回数を 16 に設定して δ =
-0.2 ~ 9.8 ppm の領域で測定した。
5.2.5.5
X 線結晶構造解析
回折データの測定には一辺が 0.1 ~ 0.3 mm の大きさの単結晶を用い、ガラスファイバー状
に結晶をグリースで固定し、-100 °C で測定した。格子定数は、6° < 2θ < 55°の範囲内の適当
な強度の回折点を用いて、最小二乗法により精密化を行った。
強度測定にはリガク社製 CCD 単結晶自動 X 線構造解析装置を用い、グラファイトで単色
化した Mo Kα線を X 線源とし、50 kV, 200 mA により測定した。強度が減衰する場合におい
ては decay correction による強度補正を行った。全反射データに対し Lorentz 因子及び偏光因
子の補正を加えたのち、I ≥ 2.00σ(I)の独立した反射を用いて解析を行った。
構造は重原子法により解析し、差フーリエ合成で得られなかった水素原子の座標は、結
晶水以外のものについては計算から求めた。非水素原子には異方性温度因子を適用し、更
に異常分散による補正、及び吸収補正を実行し、完全マトリックス最小二乗法で精密化を
行った。最小にした関数は、Σw(|F0|–|Fc|)2, w–1 = σ2(F0)である。原子散乱因子は International
Tables for Crystallography Vol.5を参照した。
構造解析、精密化は Crystal Structure 構造解析プログラム6により行い、計算は Windows
2000 をオペレーティングシステムにする市販のパーソナルコンピューターにて行った。
5.2.5.6
共鳴ラマン(rR)スペクトル
測 定 装 置 は 、 Ritsu Oyo Kogaku 社 製 Model MC-100DG spectrophotometer 、 Princeton
Instruments 社製 Model LN/CCD-1100-PB (Charge Coupled Device detector)を使用した。光源は、
Model 2060 Spectra Physics (Kr+)イオンレーザーを用いた。測定は励起波長を 406.7 nm、サン
プル濃度を 10 mM に調製した錯体溶液を高速回転用セルに充填し、低温条件下(-10 ~ -80 °C)
にて測定を行った。
98
5.2.5.7
ESI-mass スペクトル
測定装置は、Micromass 社製 LCT (ESI-TOF 型)質量分析装置を使用した。錯体の濃度は約
50 µM に調製し、マイクロシリンジを用いて毎秒 600 µl/h の速度で溶液をシリンジポンプに
よって噴霧した。校正は NaI を用いて行い、データは Mass Lynx Ver. 3.5 を用いて Windows NT
ワークステーション上にて処理した。
5.2.5.8
Stopped flow 分光測定
測定装置は、
UNISOKU 製の Stopped flow rapid scan 分光測定装置 RSP-1000 型を使用した。
モニター光源部はシャッター付き CW Xe アークランプ、分光器にはツェルニィターナー型
回折格子仕様の UNISOKU 製 MD200、マルチチャンネル測光部には MOS 型高感度フォト
ダイオードアレイを使用した。+20 ~ -90oC の任意の温度に調節された 1 cm 長の測定セル
内において、自動コック付き 3 液ミキシング装置を用いて反応溶液の混合を行い、極短時
間における UV-vis スペクトル変化を測定した。
5.2.5.9
サイックボルタンメトリー(CV)
測定装置は、BIOANALYTICAL Systems Model CV-1B-120 を使用した。測定は、参照電極
に Ag/Ag+、カウンター電極に Pt、作用電極にグラシーカーボンを用い、電位走査速度 50 ~
100mV/sec、最大-2.0 ~ +2.0V の範囲の任意の電位領域で測定を行った。測定試料は濃度 1mM
に調製した錯体溶液に支持電解質として 100 当量の(n-Bu)4NBF4 を添加したものを用い、測
定を行う前に 5 分程度アルゴンをバブリングして、十分にサンプル溶液中の酸素濃度を低
下させてから測定を行った。また、測定終了後、測定溶液にフェロセンを少量添加したの
ち再測定を行い、測定電位を Fc/Fc+基準で換算し直した。
5.2.5.10
ガスクロマトグラフ(GC)測定
測定装置として島津製作所製の GC-8APF を用い、検出器は FID、キャリアーガスには窒
素を用いた。解析は島津製作所製のクロマトパック C-R6A 及びクロマロパック C-R8A を
用い、最小ピーク幅を 5 秒、最小ピーク面積を 100 カウントとし、自動処置によりピーク
の面積を計算した。生成物の定量は内部標準法を用い、予め既知の量の反応生成物と内部
標準物質を用いて測定を行い、その面積比から検量線を作成し、その検量線に従って定量
した。以下にそれぞれの基質を用いた場合における分析条件を挙げる。
・ジメチルスルフィド
カラム:PEG-20M,
Chromosorb WAW DMCS、カラム長:3 m、カラム温度:180 ℃、イ
99
ンジェクション温度:220 ℃、キャリアーガス一次圧:600 kPa、キャリアーガス二次圧:
170 kPa、水素ガス圧:70 kPa、圧縮空気圧:60 kPa、内部標準物質:1-クロロナフタレン、
(保持時間)ジメチルスルフィド:1.1 min、ジメチルスルフォキシド:4.5 min、ジメチルスル
フォン:11.2 min.
・チオアニソール
カラム:PEG-20M,
Chromosorb WAW DMCS、カラム長:3 m、カラム温度:205 ℃、イ
ンジェクション温度:240 ℃、キャリアーガス一次圧:600 kPa、キャリアーガス二次圧:
230 kPa、水素ガス圧:70 kPa、圧縮空気圧:60 kPa、内部標準物質:1-クロロナフタレン、
(保持時間)チオアニソール:2.2 min、フェニルメチルスルフォキシド:12.7 min、ジメチル
スルフォン:24.5 min.
・シクロヘキセン
カラム:PEG-20M,
Chromosorb WAW DMCS、カラム長:3 m、カラム温度:110 ℃、イ
ンジェクション温度:220 ℃、キャリアーガス一次圧:600 kPa、キャリアーガス二次圧:
180 kPa、水素ガス圧:70 kPa、圧縮空気圧:60 kPa、内部標準物質:1,2-ジクロロベンゼン、
(保持時間)シクロヘキセン:1.0 min、シクロヘキセンオキシド:3.7 min、シクロヘキセノン:
13.0 min、シクロヘキセノール:14.4 min
100
5.3
5.3.1
結果
単核銅(II)錯体の構造と分光学的性質
5.3.1.1 [CuII(Lsq)(X)](ClO4)2 錯体の固体構造
(Lsq = bpba, bpipa, bpea ; X = MeOH, MeCN, H2O)
得られた単核銅(II)錯体の結晶学的パラメータを Table 5-1 に、主な結合長・結合角は前章
にて報告した Cu-BPBA 錯体も併せて Table 5-2 に、またその結晶構造の ORTEP 図を Figure
5-1 に示した。
錯体は[CuII(bpba)(MeCN)2](ClO4)2 を除き平面型の 4 配位型錯体であり、合成に用いた溶媒
に よ り エ ク ア ト リ ア ル 平 面 内 に MeOH, MeCN, H2O 分 子 が 配 位 し て い た 。
[CuII(Lsq)(H2O)](ClO4)2 錯体はアセトン溶媒中から単離したが、アセトン溶媒中では溶媒に含
まれている水分子が錯体に配位していると考えられる。得られた各結晶、粉末の元素分析
を行ったところ、どれも水分子が配位したものとその組成が一致した。錯体は大気下に長
時間放置しておくことにより、配位していた溶媒分子が大気中の水分子と置き代わったも
のと考えられる。結晶構造解析に成功したこれら平面型錯体間の構造的はよく似ており、
特筆すべき差異は見られなかった。
5.3.1.2 [CuII(Lsq)(X)](ClO4)2 錯体の溶液中での構造
(Lsq = bpba, bpipa, bpea ; X = MeOH, MeCN, H2O)
合成した CuIILsq 錯体の各溶媒中における分光学的測定を行った。メタノール、アセトニ
トリル、アセトン溶媒中の UV-vis スペクトル的特徴をそれぞれ Table 5-3 にまとめた。スペ
クトルはどれも平面型の銅(II)錯体に特徴的な d-d 吸収帯を示したことから、錯体は溶液中
においても平面構造を維持していると考えられる。d-d 吸収帯はどの錯体もメタノール<ア
セトン<アセトニトリル溶媒の順に短波長シフトが確認され、溶媒により空配位座に配位
している分子が置換していることを示唆していた。アセトン溶媒中においては先の X 線結
晶構造解析、並びに元素分析の結果より溶媒中の水分子が配位していると考えられる。ま
た前章でも述べたが、結晶状態では 5 配位であった[CuII(bpba)(MeCN)](ClO4)2 錯体も溶液状
態では他の CuIILsq 錯体と紫外可視吸収スペクトル的な特徴が類似しており、溶液状態では 4
配位型構造をとっているものと考えられる。
メタノール、アセトニトリル、アセトン溶媒中における各錯体の ESR スペクトルを測定し、
その解析結果を Table 5-3 にまとめた。各溶媒中における測定結果はどれも典型的な CuII(d9)
の square planar 型構造に由来するスペクトルであった。平面型銅(II)錯体において、算出さ
101
れる|A//|値は錯体の構造歪みと相関することが経験的に報告されている7。各錯体は配位子の
違いによりこの|A//|値に系統的な差が観測された。それぞれの|A//|値はどの溶媒を用いた場合
においても BPBA 錯体< BPIPA 錯体< BPEA 錯体の順に増加しており、溶液中における錯体
の平面性は BPBA 錯体< BPIPA 錯体< BPEA 錯体の順に増加していることが明らかとなった。
5.3.1.3 [CuII(Lsq)(X)]2+錯体の酸化還元電位測定
(Lsq = bpba, bpipa, bpea ; X = MeOH, MeCN, H2O)
アセトン、アセトニトリル溶媒中における[CuII(Lsq)(X)]2+錯体の酸化還元電位の測定結果
を Table 5-4 にまとめた。
錯体はどれも銅(II/I)に対応する準可逆的な酸化還元波を観測した。
酸化還元電位は BPEA 錯体< BPIPA 錯体< BPBA 錯体の順に高電位側にシフトしており、こ
の順に銅(I)状態を安定化していることが判明した。アルキル置換基である ethyl 基、
iso-propyl 基、tert-butyl 基の電子的効果の差異は小さく、錯体の電気化学的な特性はその立
体構造に起因していると考えられる。錯体はアルキル置換基の立体障害により、溶液中で
は BPEA 錯体< BPIPA 錯体<BPBA 錯体の順に平面からの歪みが大きくなっている。銅(II/I)
の効率的な電子移動を考えた場合、錯体は四面体構造に歪んでいることが望ましい8。従っ
て、錯体は BPEA 錯体< BPIPA 錯体< BPBA 錯体の順に銅(I)状態が安定化され、酸化還元電
位が高電位側へシフトしたと考えられる。
5.3.2 [CuII(Lsq)(MeOH)]2+錯体と N3-イオンとの反応 ; (Lsq = bpba, bpipa, bpea)
[CuII(Lsq)(MeOH)]2+錯体のメタノール溶液に 1 当量の NaN3 を添加し、UV-vis, ESR スペク
トル測定ならびに CV 測定を行った。測定結果を Table 5-5 にまとめた。
アジ化ナトリウムの添加により溶液は青色から濃緑色に変化し、UV-vis スペクトル測定
を行ったところ、平面型の銅(II)錯体に特徴的なスペクトルを示した。N3-→Cu(II)に対する
LMCT は BPBA, BPIPA, BPEA 錯体でそれぞれλmax = 396 nm (ε = 2870 M-1cm-1), 393 nm (ε =
2915 M-1cm-1), 390 nm (ε = 3020 M-1cm-1)に観測され、僅かではあるが系統的な短波長シフト
とモル吸光係数の増加が確認された。ESR スペクトルを測定したところ平面型の銅(II)錯体
に特徴的な形状であり、算出された各種パラメータは錯体間において特筆すべき差異が見
られなかった。錯体の|A//|値は BPBA, BPIPA, BPEA 錯体でそれぞれ 177, 178, 175 Gauss であ
り、錯体間において系統的な差異は見られず、どの錯体も溶液中で非常に高い平面性を保
持していた。錯体はエクアトリアル平面に MeOH, MeCN, H2O 等の電気的に中性な配位子を
伴った場合、その|A//|値は BPBA 錯体< BPIPA 錯体< BPEA 錯体の順に増加傾向がみられた
が、アニオン性の配位子である N3-が配位することにより錯体間の平面構造の差異が減少し、
|A//|値の差が小さくなったと考えられる。錯体の酸化還元電位はアニオン性の配位子である
102
アザイドイオンが配位することにより、中性分子である MeOH, MeCN, H2O が配位した場合
より低電位シフトしており、全てのアザイド錯体は高原子価状態が安定化されていた。さ
らに、錯体間の酸化還元電位は BPEA 錯体< BPIPA 錯体< BPBA 錯体の順に高電位側へのシ
フトが観測され、アルキル置換基の違いにより錯体の酸化還元電位に差異が見られた。
BPIPA, BPEA 錯体において得られた X 線結晶構造の結晶学的パラメータを Table 5-6、主な
結合長と結合角は前章にて報告した Lsq = BPBA も併せて Table 5-7 に、また結晶の ORTEP
図を Figure 5-2 に示した。錯体は空配位座にアザイドが一分子配位した単核の平面 4 配位型
錯体であり、錯体間の構造に大きな差異は見られなかった。各錯体の∠N(2)-Cu(1)-N(3)に大
きな差は見られなかったが、∠N(1)-Cu(1)-N(4)を比較したところ BPBA, BPIPA, BPEA 錯体
でそれぞれ 159.1(5) o, 161.03 o, 175.15o であり、結晶状態において錯体の平面性に差異が観測
された。
5.3.3 [CuII(Lsq)(MeOH)]2+錯体と Cl-イオンとの反応 ; (Lsq = bpba, bpipa, bpea)
[CuII(Lsq)(MeOH)]2+錯体のメタノール溶液に 1 当量の KCl を添加し、UV-vis, ESR スペクト
ル測定ならびに CV 測定を行った。測定結果を Table 5-5 にまとめた。
KCl の添加により目視による溶液の色変化はみられず、この青色溶液の UV-vis スペクトル
は平面型の銅(II)錯体に特徴的な吸収を示した。Cl-→Cu(II)に対する LMCT は BPBA, BPIPA,
BPEA 錯体でそれぞれλmax = 294 nm (ε = 2650 M-1cm-1), 291 nm (ε = 2970 M-1cm-1), 287 nm (ε =
3050 M-1cm-1)に観測され、僅かではあるが系統的な短波長シフトとモル吸光係数の増加が確
認された。ESR スペクトルを測定したところスペクトルは平面型の銅(II)錯体に特徴的な形
状であった。算出された g//, g⊥の値には特筆すべき差異は観測されず、|A//|値は BPBA, BPIPA,
BPEA 錯体でそれぞれ 158, 174, 177 Gauss であり、錯体の平面性には系統的な差異が見られ、
特に[CuII(bpba)(Cl-)]+錯体は他の錯体に比べ平面構造からの歪みが大きいことが判明した。
CV 測定において得られた酸化還元電位は、アニオン性配位子である塩化物イオンが配位す
ることにより、中性分子である MeOH, MeCN, H2O が配位した場合より酸化還元電位が低電
位シフトしており、全体的に高原子価状態が安定化されていた。また、錯体間において BPBA
錯体< BPIPA 錯体< BPBA 錯体の順に低電位側にシフトが観測され、アルキル置換基の違い
により錯体の酸化還元電位に差異が見られた。BPIPA, BPEA 錯体において得られた X 線結
晶構造の結晶学的パラメータを Table 5-8、主な結合長と結合角を Table 5-9 に、結晶の ORTEP
図を Figure 5-3 に示した。錯体はエクアトリアル平面に塩化物イオンが一分子配位した単核
の平面 4 配位型錯体であり、錯体間の構造に系統的な差異は見られなかった。
103
5.3.4 [CuII(Lsq)(X)]2+と過酸化水素との反応 (Lsq = bpba, bpipa, bpea)
前章において[CuII(bpba)(H2O)]2+錯体と過酸化水素を反応させることにより単核銅(II)-ハ
イドロパーオキソ錯体を合成し、UV-vis, ESR, rRaman, ESI-mass 等の分光学的測定による同
定を行なった。本項では新たに合成した[CuII(bpipa)(X)]2+, [CuII(bpea)(X)]2+錯体と過酸化水素
との反応を行い、Stopped Flow 法にて単核銅(II)-ハイドロパーオキソ錯体の生成を観測した。
[CuII(bpba)(X)]2+錯体についても同条件における測定を行い、錯体間でのスペクトルの比較を
行った。
Ar 雰囲気下にて調製した[CuII(bpipa)(H2O)]2+, [CuII(bpea)(H2O)]2+ 錯体のアセトン溶液(1
mM, -80 oC)に、2 当量のトリエチルアミンを添加し、さらに 10 当量の過酸化水素を添加し
たところ溶液は青色から緑色に変化した。しかしながら溶液は比較的早い時間で青色に戻
り、UV-vis スペクトルによる測定は困難であった。そこで、反応温度を厳密に制御でき、
且つ反応後のスペクトルの時間変化を追跡できる Stopped flow 法を用いた測定を行なった。
測定手順は 5.2.3 項に従って行ない、[CuII(Lsq)(X)]2+錯体と過酸化水素との反応の時間変化を
観測した。10 oC, アセトン溶媒中での反応により得られたスペクトルを Figure 5-4 に示した。
[CuII(bpba)(H2O)]2+錯体と過酸化水素の反応ではλmax = 350 nm (ε = 3400 M-1cm-1)に特徴的な
吸収を観測し、そのスペクトル的特徴は前章にて報告した[CuII(bpba)(OOH)]+錯体と一致し
た。また、[CuII(bpipa)(H2O)]2+錯体と過酸化水素の反応ではλmax = 388 nm (ε = 1700 M-1cm-1)
に特徴的な吸収を、[CuII(bpea)(H2O)]2+錯体と過酸化水素の反応ではλmax = 385 nm (ε = 2000
M-1cm-1)に特徴的な吸収を示した。これらはハイドロパーオキソイオンから銅(II)イオンに対
する LMCT と考えられるが、その値は[CuII(bpba)(OOH)]+錯体で観測された値より長波長シ
フトしていた。-40 oC, アセトニトリル溶媒中でも反応を行い、得られたスペクトルを Figure
5-5 に示した。[CuII(bpba)(MeCN)]2+錯体と過酸化水素の反応ではλmax = 352 nm (ε = 4200
M-1cm-1) に 特 徴 的 な 吸 収 を 観 測 し た 、 そ の ス ペ ク ト ル 的 特 徴 は 前 章 に て 報 告 し た
[CuII(bpba)(OOH)]+ 錯体とほぼ一致するが、モル吸光係数の増加が観測された。また、
[CuII(bpipa)(MeCN)]2+錯体と過酸化水素の反応ではλmax = 374 nm (ε = 1350 M-1cm-1)に特徴的
な吸収を、[CuII(bpea)(MeCN)]2+錯体と過酸化水素の反応ではλmax = 380 nm (ε = 1780 M-1cm-1)
に特徴的な吸収を示した。これらはハイドロパーオキソイオンから銅(II)イオンに対する
LMCT と考えられるが、その値は[CuII(bpba)(OOH)]+錯体に観測される値より長波長シフト
しており、またモル吸光係数の減少が見られた。
これらのスペクトル的特徴より[CuII(bpipa)(X)]2+, [CuII(bpea)(X)]2+錯体と過酸化水素との
反 応 に お い て も 、 単 核 の 銅 (II)- ハ イ ド ロ パ ー オ キ ソ 種 で あ る [CuII(bpipa)(OOH)]+,
[CuII(bpea)(OOH)]+錯体が生成していると推定される。しかしながら、これら錯体の分解は
非常に早く、他の分光学的測定は困難であった。
104
5.3.5
[CuII(Lsq)(OOH)]2+錯体の熱力学的安定性 ; (Lsq =bpba, bpipa, bpea)
Stopped flow 測定において観測された単核銅(II)-ハイドロパーオキソ錯体に特徴的な
LMCT を追跡することにより、錯体の生成・分解速度を比較した。
-40 oC アセトン溶媒中で測定した各錯体の銅(II)-ハイドロパーオキソ錯体の LMCT の時間
変化を Figure 5-6 (left column)に示した。生成速度は一次の速度式に従い、錯体の初期濃度に
は依存せず、過酸化水素濃度に依存して加速された(Figure 5-6 (right column))。生成速度は
[CuII(bpba)(OOH)]2+, [CuII(bpipa)(OOH)]2+, [CuII(bpea)(OOH)]2+錯体でそれぞれ kobs = 0.445 s-1,
2.01 s-1, 16.0 s-1 であり、錯体間において大きな差が見られた。同条件での各錯体の LMCT の
減衰を Figure 5-7 (left column)に示した。錯体の分解速度もまた一次の速度式に従うが、その
分解プロセスは錯体の初期濃度により僅かに加速され、過酸化水素の初期濃度にも依存す
ることが判明した。これは錯体の分解により生成するラジカル種は反応性の高い活性種で
あるため、生成機構に比べその過程が複雑になることに起因する。算出された分解速度定
数はそれぞれ kobs’ = 4.89×10-3 s-1, 5.58×10-2 s-1, 1.20×10-1 s-1 であり、分解速度においても錯
体間で大きな差が認められた(Figure 5-7 (right column))。
-40 oC アセトニトリル溶媒中で測定した各錯体の銅(II)-ハイドロパーオキソ錯体の LMCT
の時間変化を Figure 5-8 (left column)に示した。生成速度は一次の速度式に従い、錯体の初期
濃度に依存せず、過酸化水素濃度に依存し加速された(Figure 5-8 (right column))。生成速度は
[CuII(bpba)(OOH)]2+, [CuII(bpipa)(OOH)]2+, [CuII(bpea)(OOH)]2+錯体でそれぞれ kobs = 1.13 s-1,
8.99 s-1, 110 s-1 であった。アセトニトリル溶媒中における各錯体の生成速度はアセトン溶媒
の場合に比べて大きく、また錯体間における生成速度の差も大きく観測された。同条件で
の各錯体の LMCT の減衰を Figure 5-9 (left column)に示した。アセトニトリル溶媒中におい
ても錯体の分解プロセスは錯体の初期濃度ならびに、過酸化水素の初期濃度に依存し、ア
セトン溶媒と同様に生成するラジカル種が溶液中の水や過酸化水素、錯体自身と相互作用
していることが推測された。算出された分解速度定数はそれぞれ kobs’ = 2.29×10-2 s-1, 8.46×
10-1 s-1, 2.88 s-1 であり、錯体間で大きな差が観測された。また、生成速度の場合と同様に、
アセトニトリル溶媒中における各錯体の分解速度はアセトン溶媒の場合に比べて大きく観
測された(Figure 5-9 (right column))。
5.3.6
[CuII(Lsq)(OOH)]2+錯体による触媒反応
前章では、[CuII(bpba)(OOH)]+錯体による基質の非触媒的な酸化反応を行い、活性種の厳
密な反応特性を明らかにすると共に、反応条件を最適化することにより触媒反応の進行を
確 認 し た 。 し か し な が ら 、 本 章 に お い て 新 た に 合 成 し た [CuII(bpipa)(OOH)]+,
[CuII(bpea)(OOH)]+錯体は、[CuII(bpba)(OOH)]+錯体に比べ低温条件下でもその安定性が低く、
105
前章で行った非触媒的な酸化反応特性の検討は困難であった。そこで、これら錯体におい
ても触媒条件下で各種基質との反応を試み、 [CuII(bpba)(OOH)]+錯体も含めその反応性を比
較した。
チオアニソール、シクロヘキセンを基質とした場合の酸化反応特性を Table 5-10 にまとめ
た。アルゴン雰囲気下で調製した反応溶液(錯体:トリエチルアミン:基質 = 1 : 2 : 500 /
MeCN)を 0 oC に冷却し撹拌しながら、
錯体に対し 100 当量の過酸化水素を添加したところ、
[CuII(bpipa)(MeCN)]2+, [CuII(bpea)(MeCN)]2+ 錯体においても[CuII(bpba)(MeCN)]2+ 錯体の場合
と同様に溶液は素早く緑色を帯び、すぐに青色に戻ることを目視で確認した。このことは、
過酸化水素の添加により銅(II)-ハイドロパーオキソ錯体が生成していることが示唆してい
る。チオアニソールを基質とした触媒反応を行ったところ、各錯体の反応性には非常に大
きな差が見られた。[CuII(bpba)(OOH)]+, [CuII(bpipa)(OOH)]+, [CuII(bpea)(OOH)]+錯体の反応性
はそれぞれ TON = 100, 39, 5 であり、反応性は BPBA 錯体> BPIPA 錯体> BPEA 錯体の順に
低下した。シクロヘキセンを基質とした場合、酸化生成物であるシクロヘキセノールの収
率は[CuII(bpba)(OOH)]+, [CuII(bpipa)(OOH)]+, [CuII(bpea)(OOH)]+錯体において TON = 12, 3, 3、
シクロヘキセノンの収率は TON = 1.6, 0.8, 0.8 であり、その反応性は BPBA 錯体> BPIPA 錯
体= BPEA 錯体の順に低下していた。前章で、[CuII(bpba)(OOH)]+錯体は反応条件を最適化す
ることによりチオアニソールに対して TON = 470 まで反応性が向上したが、これは主に錯
体に対し基質の濃度が上昇した効果が大きいと考えられる。そこで基質の濃度が反応収率
に及ばす影響を検討するため、基質であるチオアニソール 100~2000 当量に変化させて各
錯 体 を 用 い た 触 媒 的 な 酸 化 反 応 を 行 っ た (Figure 5-10) 。 [CuII(bpipa)(OOH)]+,
[CuII(bpea)(OOH)]+ 錯体は基質濃度が低い条件でその反応性が著しく低下したのに対し、
[CuII(bpba)(OOH)]+錯体は基質濃度が低い場合でも 90%の収率を示した。[CuII(bpba)(OOH)]+
錯体は基質に対する選択性が他の錯体よりも高いことが判明した。[CuII(bpipa)(OOH)]+,
[CuII(bpea)(OOH)]+錯体の場合も、基質濃度の上昇に伴い反応収率の上昇が確認された。特
に、[CuII(bpipa)(OOH)]+錯体の反応性は[CuII(bpea)(OOH)]+錯体に比べ基質濃度依存性が高く、
基質の濃度の増加に伴い収率 80%に達した。
106
5.4
5.4.1
考察
アルキルアミン置換基と錯体の基本的物性の関係
配位子のアルキルアミン置換基が銅(II)錯体の立体構造に及ぼす影響を比較した。合成し
た単核銅(II)錯体はどれも 3 座型のポリピリジルアミン配位子を用いており、銅(II)イオンの
d9 型電子配置に多く見られる平面型の錯体であった。従って一座存在する平面の空配位座
には錯体の合成に用いた溶媒分子が配位していた。得られた結晶は
[CuII(bpba)(MeCN)2](ClO4)2 を除き全て 4 配位型の単核錯体であり、結晶状態において錯体間
の構造的な差異は僅かであった。平面の空配位座にアセトニトリルを伴った各錯体の結晶
構造を比較したところ、配位子に BPBA を用いた場合は 5 配位型構造であったのに対し、
BPIPA, BPEA を用いた場合は三級アミン(N(1))-銅(Cu(1))-アセトニトリル窒素(N(4))間の角
度∠N(1)-Cu(1)-N(4)はそれぞれ 153.88 o, 176.92
o
であり、錯体の平面性は BPBA 錯体 <
BPIPA 錯体 < BPEA 錯体の順に増していた。
各溶液中での分光学的測定より、錯体は溶液中においても square-planar 構造を維持してい
ることが示唆された。また、結晶状態において 5 配位型であった[CuII(bpba)(MeCN)2](ClO4)2
錯体も溶液中では 4 配位型であることは前章にて述べた。錯体間の構造的な差異は、ESR
パラメータにおいて平面型の銅(II)錯体の平面性を表す指標となる|A//|値に顕著に現れた。メ
タノール、アセトン溶媒中において錯体の|A//|値は BPBA 錯体 < BPIPA 錯体 < BPEA 錯体
の順に増加しており、これは溶液内での錯体の平面性が BPBA 錯体 < BPIPA 錯体 < BPEA
錯体の順に高くなっていることを表している。アセトニトリル溶媒を用いた場合、サンプ
ルの凍結状態が ESR 測定に適当でなかったため、スペクトルのブロード化により|A//|値の算
出は困難であった。しかしながら、先に述べた結晶構造と合わせ、アセトニトリル溶媒中
においても他の場合と同様の傾向であると推測される。このように合成した単核銅(II)錯体
はその基本骨格は同じであるが、溶液状態でその平面構造に系統的な差が観測された。こ
のエクアトリアル平面の歪みとアルキルアミンの立体障害には相関が見られ、置換基の立
体的反発により錯体平面に歪みが生じていると考えられる。このような置換基効果は錯体
の酸化還元電位にも影響を及ぼしており、BPBA 錯体 < BPIPA 錯体 < BPEA 錯体の順に高
電位シフトが観測され、錯体の高原子価状態が安定化されていた。これは溶液中での錯体
構造を反映しており、構造歪みの大きい錯体程 Cu(I)への還元反応が有利になり酸化還元電
位がシフトしたと考えられる。このように、置換基の立体障害の僅かな違いが、錯体の構
造のみならずその電気化学的特性にも大きな影響を及ぼすことが明らかとなった。
107
5.4.2
各銅(II)錯体と過酸化水素との反応
Stopped flow 法を用い[CuII(Lsq)(X)]2+錯体と過酸化水素との反応を行い、その UV-vis スペ
クトル変化を観測したところ、アセトン溶液中では CuII-BPBA, BPIPA, BPEA 錯体でそれぞ
れλmax = 350 nm (ε = 3400 M-1cm-1), 388 nm (ε = 1700 M-1cm-1), 385 nm (ε = 2000 M-1cm-1) に特
徴的な吸収を示し、アセトニトリル中ではλmax = 352 nm (ε = 4200 M-1cm-1), 374 nm (ε = 1350
M-1cm-1), 380 nm (ε = 1780 M-1cm-1)に特徴的な吸収を示した。これらはハイドロパーオキソイ
オンから CuII イオンに対する LMCT と帰属され、単核の銅(II)-ハイドロパーオキソ錯体が生
成したと考えられる。[CuII(bpipa)(OOH)]+, [CuII(bpea)(OOH)]+錯体の場合、観測された LMCT
は[CuII(bpba)(OOH)]+錯体に観測される値と比較して長波長シフト・モル吸光係数の減少が
観測されたが、その極大吸収波長は過去に報告された単核銅(II)-ハイドロパーオキソ錯体の
特徴と一致した 2。
各銅(II)錯体と過酸化水素との反応の生成・分解過程を追跡したところ、-40 oC、アセトン
溶液中では[CuII(bpba)(OOH)]+, [CuII(bpipa)(OOH)]+, [CuII(bpea)(OOH)]+錯体の生成速度は擬
一次の速度式に従い、それぞれの見かけの速度定数(kobs)は 0.445 s-1, 2.01 s-1, 16.0 s-1 であった。
また、分解速度定数も一次の速度式に従い、それぞれ kobs’ = 4.89×10-3 s-1, 5.58×10-2 s-1, 1.20
×10-1 s-1 であった。-40 oC、アセトニトリル溶液中で測定したところ生成速度は擬一次の速
度式に従い、それぞれの見かけの速度定数(kobs)は 1.13 s-1, 8.99 s-1, 110 s-1 であり、また分解
速度も一次の速度式に従いそれぞれ kobs’ = 2.29×10-2 s-1, 8.46×10-1 s-1, 2.88 s-1 であった。ア
セトニトリル溶媒中における生成・分解速度はアセトン溶媒中よりも速く、また、どちら
の溶媒中でも生成・分解速度は BPBA 錯体< BPIPA 錯体 < BPEA 錯体の順に速くなる傾向
が見られた。これは、ハイドロパーオキソ錯体の生成過程では置換基の立体障害が少ない
方が中心金属と過酸化水素との接触が容易であり、反応が進行しやすくなるためと考えら
れる。また、錯体の分解過程は生成過程に比べ複雑な要因を含んでおり、その原因を一概
に論ずることは出来ない。前章より、ハイドロパーオキソ錯体の分解速度はその錯体構造
に大きく影響されるこが明らかとなっており、4 配位型錯体間でも錯体構造がその分解速度
に影響を与えていると考えられる。溶液中の錯体構造に基づき、錯体の平面性が高い程 Cu-O
間結合が強くなるため、分解速度は BPBA 錯体 < BPIPA 錯体 < BPEA 錯体の順に早くなっ
たと推定される。錯体の分解機構を複雑にしている要因は、その分解により生成するラジ
カル種の存在である。反応性を有するこのような化学種は錯体の分解反応を促すことが予
想され、僅かではあるが分解速度は錯体の初期濃度に対して依存性を示した。さらに、錯
体の分解速度が過酸化水素濃度に対しても依存性を示したことから、発生するラジカル種
は系中に存在する過酸化水素や水分子と連鎖的に反応を起こしていると考えられる。
108
5.4.3
各錯体の反応性の比較
錯体の構造や、単核銅(II)-ハイドロパーオキソ錯体の分解過程がその酸化反応特性に及ぼ
す影響を知るため、各錯体の触媒条件下での反応性を比較した。
錯体の反応性を比較するため、基質にチオアニソール、シクロヘキセンを用いた酸化反
応を行ったところ、その反応性は錯体間で大きく異なっていた。チオアニソールを基質と
した場合、3 錯体の反応性は BPBA 錯体 > BPIPA 錯体 > BPEA 錯体の順に低下しており、
添加した酸化剤に対する収率はそれぞれ 100%, 39%, 5%であった。また、シクロヘキセンを
基質とした場合の反応性は BPBA 錯体 > BPIPA 錯体 = BPEA 錯体であり、添加した酸化剤
に対する収率はそれぞれ約 15%, 5%, 5%であった。反応の主生成物は錯体間で相違はなく、
同種の反応が進行していることが示唆された。一般的に反応中間体は不安定である程その
反応性が高いと認識されているが、本系の場合、ハイドロパーオキソ錯体の安定性は BPBA
錯体 > BPIPA 錯体 > BPEA 錯体の順であり、分解速度が遅い錯体の反応性が高かった。こ
れは、系が触媒系であることと、錯体の見かけの反応性はその失活速度に依存することに
起因している。すなわち触媒反応性の低い錯体ほど、反応開始からの錯体の失活が早いと
考えられる。また、錯体の反応性は基質の濃度に対する依存性を示した。すなわち、基質
の濃度が大きくなる程、発生するラジカル種との反応効率が増したと考えられる。これは、
同時にラジカルによる錯体の自己分解を防ぐことが予想され、反応性が向上したと考えら
れる。
興味深いことに、反応の基質濃度依存性は錯体間で大きく異なっていた。Figure 5-10 より、
最も反応性の高い BPBA 錯体は基質濃度が低い場合も失活することなく、触媒反応を進行
していることがわかる。これは、反応系中で基質を選択的に認識している可能性を示して
いる。グラフの中でこの傾向が顕著に表れているのが BPIPA 錯体であり、基質濃度に対し
て曲線的な反応収率の向上を示した。錯体は反応と同時に失活も始まるため厳密ではない
が、Figure 5-10 の縦軸を単位時間あたりの反応速度と見なした場合、ミカエリス-メンテン
型の挙動を思わせる。さらに、BPEA 錯体につても基質濃度依存性を示したがその依存度は
低く、直線的な収率の向上を示した。反応収率の基質濃度依存性は平面構造からの歪みの
大きい錯体ほど強く、錯体の構造に起因していると考えられる。すなわち、四面体構造に
歪んだ 4 配位型錯体は、5 座目の空配位座が基質の捕捉部位として働いている可能性を示唆
している。このような 5 配位型の錯体は[CuII(bpba)(MeCN)2](ClO4)2 錯体の結晶が得られてお
り、配位性の基質であるチオアニソール S 原子が錯体の空配位座と相互作用したと推定さ
れる。
109
5.5
結論
錯体構造が単核銅(II)-ハイドロパーオキソ錯体の性質に及ぼす影響を比較するため、異な
るアルキル置換基を持つ 3 座型配位子を用いた平面型の銅(II)錯体を合成した。これらの錯
体はアルキル置換基の立体障害の大きさにより結晶中、ならびに溶液中で異なった平面構
造を有していることが判明した。錯体の平面性は BPBA 錯体 < BPIPA 錯体 < BPEA 錯体の
順に増加しており、特に平面構造が歪んでいる CuII-BPBA 錯体は結晶状態においてアセト
ニトリルが二分子配位した 5 配位型の錯体が単離された。また、錯体の平面構造の差異は、
その酸化還元電位にも大きく影響を及ぼすことが判明した。このようにアルキル置換基の
僅かな違いによる錯体構造の変化が、錯体の立体構造や電気化学的性質に大きな影響を及
ぼすことが明らかとなった。これらの銅(II)錯体と過酸化水素の反応を行なったところ、生
成した銅(II)-ハイドロパーオキソ錯体の性質も大きく異なった。本章において、新たに合成
された[CuII(bpipa)(OOH)]+, [CuII(bpea)(OOH)]+錯体は単核銅(II)-ハイドロパーオキソ錯体に
見られる特徴的な LMCT が、[CuII(bpba)(OOH)]+錯体より長波長シフトしており、モル吸光
係数も減少傾向が見られた。残念ながら新たに合成したハイドロパーオキソ錯体はその早
い分解速度のため UV-vis スペクトル以外の分光学的測定は困難であり、溶液中の構造を直
接同定することは出来なかったが、その性質は出発の単核銅(II)錯体の影響を強く受けてい
ることが推定される。また、単核銅(II)-ハイドロパーオキソ錯体の生成と分解速度にも大き
な差が観測され、各錯体の生成と分解速度定数はともに BPBA 錯体 < BPIPA 錯体 < BPEA
錯体の順に大きくなっていた。さらにこれら錯体の触媒活性も各錯体で大きく異なり、そ
の反応性は BPBA 錯体 > BPIPA 錯体 > BPEA 錯体の順に減少していた。
本章では、アルキル置換基の立体障害により異なった平面性を有する一連の 4 配位型の
単核銅(II)-ハイドロパーオキソ錯体を合成し、その構造と反応性の比較を行った。結果、溶
液中で最も歪みの大きい[CuII(bpba)(OOH)]+錯体の反応性が最も高く、平面性の高い構造で
ある[CuII(bpea)(OOH)]+錯体の反応性が最も低かった。酵素 PHM の結晶構造では、CuB サイ
トは歪んだ 4 配位構造であることが明らかとなっており、反応過程で 4 ~ 5 配位型の構造を
柔軟に変化させることが指摘されている 4。実験結果も含め、このような単核銅(II)-ハイド
ロパーオキソ錯体が高い触媒活性を発現するためには、歪んだ 4 配位構造であることが重
要であることが明らかとなった。
110
References
1
(a) S. M. Miller and J. P. Klinman, Biochemistry, 1983, 22, 3091-3096. S. M. Miller and J. P.
Klinman, Biochemistry, 1985, 24, 2114-2127. (b) G. Tian, J. A. Berry and J. P. Klinman,
Biochemistry, 1994, 33, 226-234. (c) W. A. Francisco, D. J. Merkler, N. J. Blackburn and J. P.
Klinman, Biochemistry, 1998, 37, 8244-8252.
2
(a) S. Yamaguchi, S. Nagatomo, T. Kitagawa, Y. Funahashi, T. Ozawa, K. Jitsukawa and H.
Masuda, Inorg Chem, 2003, 42, 6969-6970. (b) S. Yamaguchi, A. Wada, S. Nagatomo, T.
Kitagawa, K. Jitsukawa and H. Masuda, Chem Lett, 2004, 33, 1556-1557. (c) S. Yamaguchi, A.
Kumagai, S. Nagatomo, T. Kitagawa, Y. Funahashi, T. Ozawa, K. Jitsukawa and H. Masuda,
Bull. Chem. Soc. Jpn., 2005, 78, 116-124. (d) A. Wada, M. Harata, K. Hasegawa, K. Jitsukawa,
H. Masuda, M. Mukai, T. Kitagawa and H. Einaga, Angew. Chem. Int. Ed., 1998, 37, 798-800.
(e) P. Chen, K. Fujisawa and E. I. Solomon, J. Am. Chem. Soc., 2000, 122, 10177-10193. (f) M.
Kodera, T. Kita, I. Miura, N. Nakayama, T. Kawata, K. Kano and S. Hirota, J. Am. Chem. Soc.,
2001, 123, 7715-7716.
3
(a) S. T. Prigge, A. S. Kolhekar, B. A. Eipper, R. E. Mains and L. M. Amzel, Science, 1997, 278,
1300-1305. (b) S. T. Prigge, B. A. Eipper, R. E. Mains and L. M. Amzel, Science, 2004, 304,
864-867.
4
P. Chen and E. Solomon, J. Am. Chem. Soc., 2004, 126, 4991-5000.
5
International Tables for X-ray Crystallography, Ibers, J. A.; Hamilton, W. C. eds., Kynoch Press,
Birmingham, U. K., 1974, Vol. IV.
6
“Crystal Structure” analysis program (ver 3.7) Produced by Rigaku, 2005.
7
(a) 化学総説, No.20, p179, 学芸出版センター, 東京, 1978. (b) 桜井
弘, ESR スペクト
ルの実際, p132, 廣川書店, 1989.
8
(a) 増田秀樹, 福住俊一, 「生物無機化学」, 三共出版, 2005. (b) I. Bertini, A. Sigel and H.
Sigel, Handbook on METALLOPROTEINS -Section 15-, Marcel Dekker, Inc., New York, 2001.
111
N
N
N
N
N
N
N
N
BPBA
BPIPA
N
BPEA
Chart 5-1 Ligands used in this work.
Table 5-1 Crystallographic data and refinement parameters for [CuII(Lsq)(X)](ClO4)2 (Lsq = bpipa,
bpea. X = MeOH, MeCN, H2O).
Empirical formula
Formula mass
Crystal system
Space group
a [Å]
b [Å]
c [Å]
o
α[ ]
o
β[]
o
γ[ ]
V [Å3]
Z
D calced . [g cm-3]
F (000)
µ [cm-1]
λ [Å]
T [K]
No. of refls. Measured
No. of refls. Used [I > 2σ(I 0)]
No. of Variables
R 1 [a]/ R w [b]
GOF
Empirical formula
Formula mass
Crystal system
Space group
a [Å]
b [Å]
c [Å]
o
α[ ]
o
β[]
o
γ[ ]
3
V [Å ]
Z
-3
D calced . [g cm ]
F (000)
-1
µ [cm ]
λ [Å]
T [K]
No. of refls. Measured
No. of refls. Used [I > 2σ(I 0)]
No. of Variables
[a]
[b]
R1 / Rw
GOF
[Cu(bpipa)(MeOH)](ClO4)2・MeOH
C17H25Cl2CuN3O10
565.85
triclinic
P -1 (#2)
8.648(2)
9.418(2)
14.599(4)
100.395(4)
94.025(3)
100.125(3)
1144.9(5)
2
1.641
582.00
12.453
0.71070
-100.0
8920
3897
323
0.0407 / 0.1074
1.008
[Cu(bpipa)(MeCN)](ClO4)2
C17H22Cl2CuN4O8
544.83
monoclinic
P 21/n (#14)
8.408(16)
13.51(3)
19.78(4)
101.244(19)
2204(8)
4
1.642
1116.00
12.854
0.71070
-100.0
17040
14884
311
0.0455 / 0.1607
1.004
[Cu(bpea)(MeOH)](ClO4)2・MeOH [Cu(bpea)(MeCN)](ClO4)2・2H2O
C16H24Cl2CuN4O10
C16H24Cl2CuN3O10
552.83
566.84
triclinic
monoclinic
P 21/n (#14)
P -1 (#2)
8.387(8)
16.050(5)
9.479(19)
7.283(2)
14.400(14)
20.109(6)
99.76(6)
91.567(2)
89.100(4)
98.83(5)
1113(3)
2350.3(12)
2
4
1.649
1.602
568.00
1164.00
12.786
14.50
0.71070
0.71070
-100.0
-100.0
8844
18101
4399
4479
313
309
0.0437 / 0.1429
0.080/0.147
1.000
1.19
112
[Cu(bpea)(H2O)](ClO4)2
C14H19Cl2CuN3O9
507.77
orthorhombic
P 212121 (#19)
8.336(7)
14.370(14)
16.106(14)
1929(3)
4
1.748
1036
14.633
0.71070
-100.0
15307
14681
281
0.0625 / 0.2113
1.000
Table 5-2 Selected bond lengths and angles of [CuII(Lsq)(X)](ClO4)2 (Lsq = bpba, bpipa, bpea, X =
MeOH, MeCN, H2O).
[Cu(bpba)(MeOH)](ClO4)2
Cu(1)-N(1)
2.064(2)
Cu(1)-N(2)
1.936(3)
Cu(1)-N(3)
1.964(3)
Cu(1)-O(1)
2.017(2)
[Cu(bpipa)(MeOH)](ClO4)2
Cu(1)-N(1)
2.063(2)
Cu(1)-N(2)
1.958(2)
Cu(1)-N(3)
1.954(2)
Cu(1)-O(1)
1.989(2)
[Cu(bpea)(MeOH)](ClO4)2
Cu(1)-N(1)
2.042(2)
Cu(1)-N(2)
1.962(2)
Cu(1)-N(3)
1.971(2)
Cu(1)-O(1)
1.9863(18)
N(1)-Cu(1)-O(1)
N(2)-Cu(1)-N(3)
N(1)-Cu(1)-N(2)
N(1)-Cu(1)-N(3)
N(2)-Cu(1)-O(1)
N(3)-Cu(1)-O(1)
N(1)-Cu(1)-O(1)
N(2)-Cu(1)-N(3)
N(1)-Cu(1)-N(2)
N(1)-Cu(1)-N(3)
N(2)-Cu(1)-O(1)
N(3)-Cu(1)-O(1)
N(1)-Cu(1)-O(1)
N(2)-Cu(1)-N(3)
N(1)-Cu(1)-N(2)
N(1)-Cu(1)-N(3)
N(2)-Cu(1)-O(1)
N(3)-Cu(1)-O(1)
179.68(10)
167.8(1)
84.01(10)
85.1(1)
96.0(1)
94.9(1)
175.23(10)
167.71(10)
84.46(9)
83.87(10)
96.03(9)
95.29(10)
179.06(8)
167.07(9)
84.10(8)
83.40(8)
95.15(9)
97.32(8)
[Cu(bpba)(MeCN)2](ClO4)2
Cu(1)-N(1)
2.085(3)
Cu(1)-N(2)
1.955(2)
Cu(1)-N(3)
1.961(2)
Cu(1)-N(4)
2.019(3)
Cu(1)-N(5)
2.277(3)
[Cu(bpipa)(MeCN)](ClO4)2
Cu(1)-N(1)
2.0300(15)
Cu(1)-N(2)
1.9664(13)
Cu(1)-N(3)
1.9578(13)
Cu(1)-N(4)
2.0011(16)
[Cu(bpea)(MeCN)](ClO4)2
Cu(1)-N(1)
2.045(4)
Cu(1)-N(2)
1.963(4)
Cu(1)-N(3)
1.974(5)
Cu(1)-N(4)
1.985(5)
N(1)-Cu(1)-N(2)
N(1)-Cu(1)-N(3)
N(1)-Cu(1)-N(4)
N(1)-Cu(1)-N(5)
N(2)-Cu(1)-N(3)
N(2)-Cu(1)-N(4)
N(2)-Cu(1)-N(5)
N(3)-Cu(1)-N(4)
N(3)-Cu(1)-N(5)
N(4)-Cu(1)-N(5)
N(1)-Cu(1)-N(2)
N(1)-Cu(1)-N(3)
N(1)-Cu(1)-N(4)
N(2)-Cu(1)-N(3)
N(2)-Cu(1)-N(4)
N(3)-Cu(1)-N(4)
N(1)-Cu(1)-N(2)
N(1)-Cu(1)-N(3)
N(1)-Cu(1)-N(4)
N(2)-Cu(1)-N(3)
N(2)-Cu(1)-N(4)
N(3)-Cu(1)-N(4)
84.38(11)
84.24(11)
160.95(13)
112.92(14)
164.30(12)
93.88(13)
97.41(11)
93.34(12)
96.98(12)
86.13(15)
[Cu(bpba)(OH2)](ClO4)2
Cu(1)-N(1)
2.08(2)
Cu(1)-N(2)
1.90(2)
Cu(1)-N(3)
1.91(2)
Cu(1)-O(1)
2.03(2)
N(1)-Cu(1)-O(1)
N(2)-Cu(1)-N(3)
N(1)-Cu(1)-N(2)
N(1)-Cu(1)-N(3)
N(2)-Cu(1)-O(1)
N(3)-Cu(1)-O(1)
160.6(8)
166.6(7)
82.6(8)
84.7(7)
95.0(8)
95.6(8)
83.64(5)
84.58(5)
153.88(5)
165.05(5)
96.75(6)
97.88(5)
[Cu(bpea)(OH2)](ClO4)2
Cu(1)-N(1)
2.024(2)
Cu(1)-N(2)
1.970(2)
Cu(1)-N(3)
1.956(2)
Cu(1)-O(1)
2.016(2)
N(1)-Cu(1)-O(1)
N(2)-Cu(1)-N(3)
N(1)-Cu(1)-N(2)
N(1)-Cu(1)-N(3)
N(2)-Cu(1)-O(1)
N(3)-Cu(1)-O(1)
167.39(8)
166.68(9)
82.83(9)
83.86(10)
99.72(9)
93.36(9)
113
82.7(2)
83.4(2)
176.9(2)
166.1(2)
96.2(2)
97.7(2)
[CuII(bpipa)(MeOH)]2+
[CuII(bpea)(MeOH)]2+
[CuII(bpipa)(MeCN)]2+
[CuII(bpea)(MeCN)]2+
[CuII(bpea)(H2O)]2+
Figure 5-1 ORTEP drawing of [CuII(Lsq)(X)](ClO4)2 (Lsq = bpipa, bpea, X = MeOH, MeCN, H2O)
with the atom labeling scheme. The thermal ellipsoids are at the 50% probability level, and the
hydrogen atoms were omitted for clarity.
114
Table 5-3 Spectroscopic feature of [CuII(Lsq)(X)](ClO4)2 (Lsq = bpba, bpipa, bpea, X = MeOH,
MeCN, H2O).
d-d/ nm ( ε/ M-1cm-1)
g⊥
g//
|A//|
[Cu(bpba)(MeOH)] / MeOH
[Cu(bpipa)(MeOH)]2+ / MeOH
[Cu(bpea)(MeOH)]2+ / MeOH
650 (117)
648 (106)
654 (95)
2.27
2.26
2.26
2.07
2.06
2.06
154
165
166
[Cu(bpba)(MeCN)]2+ / MeCN
[Cu(bpipa)(MeCN)]2+ / MeCN
[Cu(bpea)(MeCN)]2+ / MeCN
612 (165)
611 (167)
605 (138)
-
-
-
[Cu(bpba)(H2O)]2+ / Acetone
complex / solvent
2+
640 (128)
2.25
2.08
153
2+
640 (112)
2.25
2.06
166
2+
648 (91)
2.26
2.06
170
[Cu(bpipa)(H2O)] / Acetone
[Cu(bpea)(H2O)] / Acetone
Table 5-4 Electrochemical data of [CuII(Lsq)(X)](ClO4)2 (Lsq = bpba, bpipa, bpea, X = MeCN, H2O).
complex / solvent
Epa [mV]
Epc [mV]
∆E [mV]
E1/2(Ag/Ag+) [mV]
E1/2(Fc/Fc+) [mV
[Cu(bpba)(MeCN)]2+ / MeCN
281
208
73
245
-99
[Cu(bpipa)(MeCN)] / MeCN
207
141
66
174
-179
[Cu(bpea)(MeCN)]2+ / MeCN
152
77
75
115
-239
[Cu(bpba)(H2O)]2+ / Acetone
119
26
93
72.5
-282
[Cu(bpipa)(H2O)] / Acetone
70
-19
89
25.5
-330
[Cu(bpea)(H2O)]2+ / Acetone
1
-78
79
-38.5
-386
2+
2+
Table 5-5 Spectroscopic and electrochemical properties for [CuII(Lsq)(X)]2+ (Lsq = bpba, bpipa, bpea,
X = Cl-, N3-).
-1
-1
UV-vis /nm (ε/M cm )
LMCT
d-d
complex
g⊥
ESR
g//
|A//|
E1/2 (Ag/Ag+)
CV /mV
E1/2 (Fc/Fc+)
∆E
[Cu(bpba)(Cl)]+
+
[Cu(bpipa)(Cl)]
+
[Cu(bpea)(Cl)]
294 (2650)
291 (2970)
287 (3050)
687 (175)
677 (153)
675 (155)
2.04
2.05
2.04
2.22
2.22
2.22
158
174
177
84.5
15.5
-19.5
87
95
93
-396
-464
-498
+
396 (2870)
393 (2915)
390 (3020)
657 (361)
657 (311)
650 (306)
2.05
2.05
2.06
2.22
2.22
2.23
177
178
175
49.5
-23.0
-67.0
85
86
88
-427
-503
-551
[Cu(bpba)(N3)]
+
[Cu(bpipa)(N3)]
+
[Cu(bpea)(N3)]
115
Table 5-6 Crystallographic data and refinement parameters for [CuII(bpipa)(N3-)]ClO4,
[CuII(bpea)(N3-)]ClO4.
Empirical formula
Formula mass
Crystal system
Space group
a [Å]
b [Å]
c [Å]
α [o]
β [o]
γ [o]
V [Å3]
Z
D calced . [g cm-3]
F (000)
µ [cm-1]
λ [Å]
T [K]
No. of refls. Measured
No. of refls. Used [I > 2σ(I 0)]
No. of Variables
R 1 [a]/ R w [b]
GOF
[Cu(bpipa)(N3)]ClO4
C15H19ClCuN6O4
446.35
orthorhombic
Pbca (#61)
15.7251(10)
14.8101(15)
15.7452(15)
3666.9(6)
8
1.617
1832.00
13.721
0.71070
-100.0
27833
2038
263
0.0298 / 0.0503
0.728
[Cu(bpea)(N3)]ClO4
C14H17ClCuN6O4
432.33
monoclinic
P 21/n (#14)
8.974(2)
14.207(2)
13.701(10)
102.057(20)
1708.2(14)
4
1.681
884.00
14.698
0.71070
-100.0
13310
11692
252
0.0466 / 0.1668
1.000
Table 5-7 Selected bond lengths and angles of [CuII(bpipa)(N3-)]+ and [CuII(bpea)(N3-)]+ together
with those of [CuII(bpba)(N3-)]+.
[Cu(bpba)(N3)]ClO4
Cu(1)-N(1)
2.071(8)
Cu(1)-N(2)
1.977(8)
Cu(1)-N(3)
1.960(8)
Cu(1)-N(4)
1.94(1)
[Cu(bpipa)(N3)]ClO4
Cu(1)-N(1)
2.045(2)
Cu(1)-N(2)
1.967(3)
Cu(1)-N(3)
1.988(3)
Cu(1)-N(4)
1.948(3)
[Cu(bpea)(N3)]ClO4
Cu(1)-N(1)
2.0570(12)
Cu(1)-N(2)
1.9852(17)
Cu(1)-N(3)
1.9745(15)
Cu(1)-N(4)
1.9501(14)
N(1)-Cu(1)-N(2)
N(1)-Cu(1)-N(3)
N(1)-Cu(1)-N(4)
N(2)-Cu(1)-N(3)
N(2)-Cu(1)-N(4)
N(3)-Cu(1)-N(4)
N(1)-Cu(1)-N(2)
N(1)-Cu(1)-N(3)
N(1)-Cu(1)-N(4)
N(2)-Cu(1)-N(3)
N(2)-Cu(1)-N(4)
N(3)-Cu(1)-N(4)
N(1)-Cu(1)-N(2)
N(1)-Cu(1)-N(3)
N(1)-Cu(1)-N(4)
N(2)-Cu(1)-N(3)
N(2)-Cu(1)-N(4)
N(3)-Cu(1)-N(4)
85.0(3)
84.6(3)
159.1(5)
164.9(4)
101.9(4)
91.7(4)
116
83.47(12)
82.37(12)
161.03(13)
165.64(12)
98.82(13)
95.35(13)
82.82(5)
82.22(5)
175.15(6)
165.00(5)
101.35(6)
93.56(6)
Figure 5-2 ORTEP drawing of [CuII(Lsq)(N3-)]ClO4 (Lsq = bpipa, bpea) with the atom labeling
scheme. The thermal ellipsoids are at the 50% probability level, and the hydrogen atoms were
omitted for clarity. (left) [CuII(bpipa)(N3-)]ClO4, (right) [CuII(bpea)(N3-)]ClO4
Table 5-8 Crystallographic data and refinement parameters for [CuII(bpipa)(Cl-)]ClO4,
[CuII(bpea)(Cl-)]ClO4.
Empirical formula
Formula mass
Crystal system
Space group
a [Å]
b [Å]
c [Å]
α [o]
β [o]
γ [o]
V [Å3]
Z
D calced . [g cm-3]
F (000)
-1
µ [cm ]
λ [Å]
T [K]
No. of refls. Measured
No. of refls. Used [I > 2σ(I 0)]
No. of Variables
R 1 [a]/ R w [b]
GOF
[Cu(bpipa)(Cl)]ClO4
C30H38Cl4Cu2N6O8
879.57
triclinic
P -1 (#2)
8.180(3)
14.70(3)
14.441(4)
90.01(11)
90.026(18)
94.76(9)
1731(4)
2
1.687
900.00
15.962
0.71070
-100.0
13924
4882
489
0.0554 / 0.1239
1.008
117
[Cu(bpea)(Cl)]ClO4
C14H17Cl2CuN3O4
425.76
monoclinic
P 21/n
8.504(10)
14.455(14)
13.65(2)
101.52(4)
1645(4)
4
1.719
868.00
16.769
0.71070
-100.0
12812
11305
231
0.0564 / 0.1967
1.003
Table 5-9 Selected bond lengths and angles of [CuII(bpipa)(Cl-)]+ and [CuII(bpea)(Cl-)]+ together
with those of [CuII(bpba)(Cl-)]+.
[Cu(bpba)(Cl)]ClO4
Cu(1)-N(1)
2.061(3)
Cu(1)-N(2)
1.965(4)
Cu(1)-N(3)
1.966(3)
Cu(1)-Cl(1)
2.2119(14)
[Cu(bpipa)(Cl)]ClO4
Cu(1)-N(1)
2.046(4)
Cu(1)-N(2)
1.983(4)
Cu(1)-N(3)
1.972(4)
Cu(1)-Cl(1)
2.2302(16)
[Cu(bpea)(Cl)]ClO4
Cu(1)-N(1)
2.0369(16)
Cu(1)-N(2)
1.973(2)
Cu(1)-N(3)
1.988(2)
Cu(1)-Cl(1)
2.2218(6)
N(1)-Cu(1)-N(2)
N(1)-Cu(1)-N(3)
N(1)-Cu(1)-Cl(1)
N(2)-Cu(1)-N(3)
N(2)-Cu(1)-Cl(1)
N(3)-Cu(1)-Cl(1)
N(1)-Cu(1)-N(2)
N(1)-Cu(1)-N(3)
N(1)-Cu(1)-Cl(1)
N(2)-Cu(1)-N(3)
N(2)-Cu(1)-Cl(1)
N(3)-Cu(1)-Cl(1)
N(1)-Cu(1)-N(2)
N(1)-Cu(1)-N(3)
N(1)-Cu(1)-Cl(1)
N(2)-Cu(1)-N(3)
N(2)-Cu(1)-Cl(1)
N(3)-Cu(1)-Cl(1)
84.32(16)
84.99(15)
152.71(13)
159.20(18)
98.82(13)
98.82(13)
84.31(18)
98.67(13)
149.77(14)
163.92(18)
97.33(14)
98.67(13)
82.56(6)
81.39(7)
173.31(4)
163.70(7)
98.42(5)
97.85(5)
Figure 5-3 ORTEP drawing of [CuII(Lsq)(Cl-)]ClO4 (Lsq = bpipa, bpea) with the atom labeling
scheme. The thermal ellipsoids are at the 50% probability level, and the hydrogen atoms were
omitted for clarity. (left) [CuII(bpipa)(Cl-)]ClO4, (right) [CuII(bpea)(Cl-)]ClO4
118
0.8
0.7
Absorbance
0.6
0.5
0.4
0.3
0.2
0.1
0
300
350
400
450
500
550
600
Wavelength [nm]
Figure 5-4 UV-vis spectra of [CuII(Lsq)(OOH)]+ complexes (0.25 mM) measured
by using stopped flow technique in acetone solution (10oC).
[CuII(bpba)(OOH)]+ (solid line), [CuII(bpipa)(OOH)]+ (bold line),
[CuII(bpea)(OOH)]+ (bold dotted line), respectively.
1.2
1
Absorbance
0.8
0.6
0.4
0.2
0
300
350
400
450
500
550
600
wavelength [nm]
Figure 5-5 UV-vis spectra of [CuII(Lsq)(OOH)]+ complexes (0.25 mM) measured
by using stopped flow technique in MeCN solution (-40oC).
[CuII(bpba)(OOH)]+ (solid line), [CuII(bpipa)(OOH)]+ (bold line),
[CuII(bpea)(OOH)]+ (bold dotted line), respectively.
119
3.5
0.9
3
ln(Ainf/(Ainf-At))
Absorbance (350 nm)
1
0.8
0.7
2.5
2
1.5
0.6
0.5
1
0
1
2
3
4
5
6
7
0.5
8
0
1
2
time [s]
0.35
5
3.5
3
0.25
ln(Ainf/(Ainf-At))
Absorbance (388 nm)
4
4
0.3
0.2
0.15
2.5
2
1.5
1
0.1
0.5
0
0.05
0
0.5
1
1.5
2
0
0.5
1
1.5
2
time [s]
time [s]
0.45
3.5
0.4
3
0.35
2.5
ln(Ainf/(Ainf-At))
Absorbance (385 nm)
3
time [s]
0.3
0.25
0.2
2
1.5
1
0.15
0.5
0.1
0
0.05
0
0.05
0.1
0.15
0.2
0.25
0.3
0
0.35
0.05
0.1
0.15
0.2
time [s]
time [s]
Figure 5-6 Representative spectral changes for generation processes of copper(II)-hydroperoxo
species in acetone at -40 oC. <left column> Time courses of the characteristic absorbance change for
bpba(top), bpipa(middle) and bpea(bottom). <right column> First-order kinetics plots for bpba(top),
bpipa(middle) and bpea(bottom).
120
0.5
0.9
0
0.8
-0.5
ln((At-Ainf )/(A0-Ainf ))
Absorbance (350 nm)
1
0.7
0.6
0.5
0.4
-1
-1.5
-2
-2.5
-3
0.3
-3.5
0.2
0
100
200
300
400
500
0
600
100
200
300
400
500
600
20
25
30
20
25
30
time [s]
time [s]
0.35
0
0.25
ln((At-Ainf)/(A0-Ainf))
Absorbance (388 nm)
0.3
0.2
0.15
-0.5
-1
0.1
0.05
0
20
40
60
80
100
-1.5
120
0
5
10
15
time [s]
0.45
0
0.4
-0.5
ln((At-Ainf)/(A0-Ainf))
Absorbance (385 nm)
time [s]
0.35
0.3
0.25
0.2
-1
-1.5
-2
-2.5
0.15
-3
0.1
0
10
20
30
40
0
50
5
10
15
time [s]
time [s]
Figure 5-7 Representative spectral changes for decomposition processes of copper(II)-hydroperoxo
species in acetone at -40 oC. <left column> Time courses for the characteristic absorbance change of
bpba(top), bpipa(middle) and bpea(bottom). <right column> First-order kinetics plots for bpba(top),
bpipa(middle) and bpea(bottom).
121
4
1.2
3.5
3
1
ln(Ainf/(Ainf-At))
Absorbance (352 nm)
1.1
0.9
0.8
2.5
2
0.7
1.5
0.6
1
0.5
0.5
0
0.5
1
1.5
2
2.5
3
0
3.5
0.5
1.5
2
2.5
3
2.5
0.4
0.35
2
0.3
ln(Ainf/(Ainf-At))
Absorbance (374 nm)
1
time [s]
time [s]
0.25
0.2
1.5
1
0.15
0.5
0.1
0.05
0
0.05
0.1
0.15
0.2
0
0.25
0
0.05
time [s]
0.15
0.2
0.25
time [s]
3.5
0.5
3
ln(Ainf/(Ainf-At))
0.45
Absorbance (380 nm)
0.1
0.4
0.35
0.3
2.5
2
1.5
1
0.5
0.25
0
0.005
0.01
0.015
0.02
0.025
0.03
0
0.005
0.01
0.015
0.02
time [s]
time [s]
Figure 5-8 Representative spectral changes for generation processes of copper(II)-hydroperoxo
species in MeCN at -40 oC. <left column> Time courses of the characteristic absorbance change for
bpba (top), bpipa (middle) and bpea (bottom). <right column> First-order kinetics plots for bpba
(top), bpipa (middle) and bpea (bottom)
122
0
1
-0.5
ln((At-Ainf )/(A0-Ainf ))
Absorbance (352 nm)
1.2
0.8
0.6
-1
-1.5
-2
0.4
0.2
-2.5
0
50
100
150
200
250
300
0
50
100
0.5
0
0.45
-0.5
0.4
-1
0.35
0.3
0.25
0.2
0.15
-2
-2.5
-3
0
0.5
1
1.5
-3.5
2
0
0.2
0.4
0.6
0.8
1
time [s]
0.5
-0.5
0.45
-1
0.4
ln((At-Ainf)/(A0-Ainf))
Absorbance (380 nm)
200
-1.5
time [s]
0.35
0.3
0.25
0.2
0.15
150
time [s]
ln((At-Ainf)/(A0-Ainf))
Absorbance (374 nm)
time [s]
-1.5
-2
-2.5
-3
-3.5
0
0.5
1
1.5
-4
2
time [s]
0
0.2
0.4
0.6
0.8
1
1.2
time [s]
Figure 5-9 Representative spectral changes for decomposition processes of copper(II)-hydroperoxo
species in acetone at -40 oC. <left column> Time courses for the characteristic absorbance change of
bpba (top), bpipa (middle) and bpea (bottom). <right column> First-order kinetics plots for bpba
(top), bpipa (middle) and bpea (bottom).
123
Table 5-10 Comparison of the catalytic reactivities among the copper(II)-hydroperoxo complexes.
Cu-BPBA
Oxidation of thioanisole
Phenylmethylsulfone (TON)
100
Cu-BPIPA
38.6
Cu-BPEA
5.3
Cu-BPBA
Oxidation of cyclohexene
cyclohexenol (TON) cyclohexenon (TON)
11.6
1.6
Cu-BPIPA
3.2
0.8
Cu-BPEA
3.2
0.8
100
TON
80
60
40
20
0
0
500
1000
1500
2000
Ph-S-Me [eq]
Figure 5-10 Correlation between catalytic reactivities and concentrations of Ph-S-Me.
[CuII(bpba)(OOH)]+ (solid line ●), [CuII(bpipa)(OOH)]+ (bold dotted line ■),
[CuII(bpea)(OOH)]+ (dotted line ▲).
124
第6章
4 座型配位子を用いた単核鉄(III)-ハイドロパーオキソ錯体
の合成とその触媒反応性の検討
6.1
序論
前章までは中心金属に銅を含有する酸化酵素 DβM, PHM のモデル化学的な反応中間体の
合成と、その酸化反応特性を明らかにした。新たに合成した平面型の銅(II)-ハイドロパーオ
キソ錯体は外部の有機基質に対する反応性を示し、反応条件の最適化により触媒反応の進
行を確認した。生態系には中心金属に鉄を含有する酸化酵素もまた数多く存在し、本研究
室ではそのような酵素を対象としたモデル化学的な検討も行ってきた。不飽和脂肪酸を酸
化するリポキシゲナーゼ1や、ナフタレンのシス-ジオール化を触媒するナフタレンジオキシ
ゲナーゼ2等の酸化酵素は、それぞれ鉄(III)-ハイドロパーオキソもしくは鉄(III)-アルキルパ
ーオキソ種を反応中間体とし基質を酸化することが提唱されている3。また、グリコペプチ
ドであるブレオマイシン4は抗ガン剤として使用され、単核鉄(III)-ハイドロパーオキソ種を
中間体として遺伝子の酸化的分解を行う。このように中心金属に鉄を有する単核鉄(III)-ハ
イドロパーオキソ(アルキルパーオキソ)種も生体内において重要な反応を触媒する活性種
であり、非常に興味深い。
モデル化学的な単核鉄(III)-ハイドロパーオキソ錯体の合成や反応特性の検討は過去に多
数報告されている。Que らは TPA 類縁体を配位子に用いた一連の錯体を合成し、ピリジン 6
位に導入した置換基により鉄(III)イオンのスピン状態が制御出来ることを報告している5。
また、合成した鉄(III)-ハイドロパーオキソ錯体は low-spin の場合、ハイドロパーオキソイ
オンの分解により FeV(=O)OH 種を形成し、この反応中間体がオレフィンに対する求電子的
な水酸化反応を行うことを報告している6。一方、high-spin 型の鉄(III)-ハイドロパーオキソ
錯体は求核的な酸素分子の挿入反応を行うことが知られているが、その詳細な機構はわか
っていない 6。どちらの場合も共通していることは、ハイドロパーオキソイオンの O-O 間結
合が開裂し次段階の活性種を生成することである。またその際に、ハイドロパーオキソイ
オンのシス位に存在している空配位座が重要な役割を果たしていることが明らかとなって
いる。
本研究室では過去に嵩高い置換基を持つ配位子 H2BPPA を用いて、high-spin 型の鉄(III)ハイドロパーオキソ錯体([FeIII(H2bppa)(OOH)]2+)を単離することに成功し、この錯体はチオ
エーテル類の酸化に対し求核的な反応性を示した7。また、配位子 MPPA (bis(2-pyridylmethyl)
(6-pivalamido-2-pyridylmethy)amine)を基本骨格として、ピリジン 4, 6 位に様々な置換基を導
125
入した一連の鉄(III)-ハイドロパーオキソ錯体を合成し、Que らと同様に置換基効果によるス
ピン状態の制御にも成功している8。
本章においては、このような研究をもとに、捕捉したハイドロパーオキソイオンの活性
化に重要と考えられるシス位の空配位座を導入した錯体の合成行った。Wada らの報告にあ
る配位子 H2BPPA は、ピリジン 6 位に存在する二つのピバルアミド酸素が鉄イオンに配位す
ることが可能であるため 6~7 座型の多座配位型の配位子である。そこで今回、Yamaguchi
らの報告した 5 配位型配位子である MPPA 類縁体を基に、一部のピリジン置換基を立体障
害 の 大 き い ア ル キ ル 置 換 基 に 変 換 し た
4
座 型 配 位 子
PPBA((6-pivalamide-2-pyridylmethyl)(2-pyridylmethyl)tert-butylamine)を合成した。配位子 PPBA
を用いて合成した鉄(II)錯体はシス位に二つの空配位座を有すると考えられ、このような錯
体を用いた単核鉄(III)-ハイドロパーオキソ錯体の合成と、各種基質に対する酸化反応実験
を行った。これによりシス位の空配位座が錯体の反応性に及ばす影響を検討した。
126
6.2
実験
6.2.1
配位子合成
第 6 章にて用いた配位子の構造を Chart 6-1 に示した。
6.2.1.1
2-pyridylmethyl-tert-butylamine の合成
tert-butylamine 22 g (0.3 mol)、2-chloromethyl-pyridine 9.84 g (0.06mol)を 1,4-ジオキサン 200
ml に溶かした。これに少量の水に溶かした KOH 6.73 g (0.12 mol)を加え TLC (展開溶媒;ク
ロロホルム:メタノール= 7 : 1) で反応の進行を確認しながら 100 oC にて約 3 時間還流した。
反応の進行に伴い KCl が析出し、反応溶液は褐色を呈した。濾過により析出した KCl を除
去したのち、減圧濃縮により暗褐色の油状混合物を得た。これをクロロホルムに溶解させ
0.1N HCl aq で中和したのち有機相を分取、飽和食塩水、無水 MgSO4 を用いて脱水処理をし
たのち、減圧濃縮してシリカゲルカラム (クロロホルム : メタノール = 100 : 0 ~ 5)により
精製を行い、
褐色油状の溶液 8.1 g を得た。Yield 83 %
1
H-NMR (CDCl3, 300MHz) ; 1.20(s, 9H),
1.85(s, 1H), 3.88(s,2H), 7.15(t, 1H), 7.33(d, 1H), 7.63(t, 1H), 8.54 (d,1H)
6.2.2.2
PPBA:(6-pivalamide-2-pyridylmethyl)(2-pyridylmethyl)tert-butylamine の合成
2-pyridylmethyl-tert-butylamine
3.56
g
(0.027
mol)
と
別
途
合
成
し
た
2-bromomethyl-6-pivalamidopyridine9 5.42 g (0.02 mol)を 1,4-ジオキサン 200 ml に溶かし、こ
れに水 20ml に溶かした KOH 1.2 g (0.022 mol)を加えた。溶液を撹拌し、TLC にて反応を追
跡しながら 50 oC にて約 5 時間反応させた。濾過により析出した KBr を除去したのち、減圧
濃縮により暗褐色の油状混合物を得た。これをクロロホルムに溶解させ 0.1N HCl aq で中和
したのち有機相を分取、飽和食塩水、無水 MgSO4 を用いて脱水処理をしたのち、減圧濃縮
してシリカゲルカラム (クロロホルム : メタノール = 100 : 0 ~ 3)により精製を行い、白色
の粉末 1.05 g を得た。Yield 15 %
1
H-NMR (CDCl3, 300MHz) ; 1.17(s, 9H), 1.30(s, 9H),
3.81(s,2H), 3.93(s, 2H), 7.00(t, 1H), 7.20(d, 1H), 7.46-7.54(m, 3H), 7.94(d, 1H), 8.37 (d,1H)
6.2.2
錯体合成
6.2.2.1 [FeII(ppba)](OTf)2 (OTf = -OSO2CF3) 錯体の合成
Ar 雰囲気下において、アセトニトリル 2 ml に加熱溶解させた FeII(OTf)2 53.1 mg (0.15
mmol)に、PPBA 53.2 mg (0.15 mmol)を加えることにより溶液は透明から黄色に変化した。こ
れにジエチルエーテルを溶液が少し白濁するまで添加し、一日放置することにより黄色板
127
状の結晶 57.8 mg を得た。結晶は高度に層状化した微結晶であり、X 線による結晶構造解析
は困難であった。Yield: 53 %. Anal. Calcd for [FeII(ppba))](OTf)2 (C23H30FeN4O7S2F6): C, 38.81;
H, 4.11; N, 7.83. Found: C, 38.99; H, 4.27; N, 7.91.
6.2.2.2 [FeII(ppba)(MeCN)2](ClO4)2 錯体の合成
Ar 雰囲気下において、[FeII(ppba)](OTf)2 錯体 21.3 mg (0.03 mmol)をアセトニトリル溶媒に
溶かし、これに NaClO4 3.67 mg (0.03 mmol)を加え、ジエチルエーテル拡散により結晶化を
行 っ た 。 得 ら れ た 黄 色 結 晶 は 二 種 類 の FeII(ppba) 錯 体 を 含 ん で お り 、 ひ と つ は
[FeII(ppba)](OTf)2 錯 体 で あ り 、 ひ と つ は [FeII(ppba)(MeCN)2](ClO4)2 錯 体 で あ っ た 。
[FeII(ppba)(MeCN)2](ClO4)2 錯体については X 線による構造解析に成功した。
6.2.3
鉄(II)錯体と過酸化水素との反応
鉄(II)錯体溶液 1.0 mM を Ar 雰囲気下にて低温測定用のセルに封入したのち、低温測定ユ
ニットと共に UV-vis 測定器に取り付けた。ドライアイス-メタノール溶媒を用いてサンプル
を-78 oC に冷却したのち、過酸化水素水 10 当量をシリンジにて添加し、撹拌することによ
り反応を行い、UV-vis スペクトル変化を観測した。また、同溶液は ESR, 共鳴ラマン測定を
行った。
6.2.4
触媒反応
鉄(II)錯体 4 µmol、基質 2 mmol、GC 用の内部標準物質をアセトンもしくはアセトニトリ
ル溶媒 2 ml に溶解させ反応容器に封入し、Ar 置換を行なった。これを ice bath にて 0 oC に
冷却後、過酸化水素水 0.4 mmol をシリンジにて添加して 15 分間攪拌することにより反応を
行なった。反応溶液はシリンジにて 50 µl 採取し、過剰量のトリフェニルホスフィン(PPh3)
を添加したのち室温まで昇温させ、ガスクロマトグラフ測定により反応生成物の定量を行
なった。過酸化水素は 15 分おきに再添加し、酸化反応の経時変化を追跡した。
6.2.5
6.2.5.1
測定装置
紫外可視吸収(UV-vis)スペクトル
測定装置は、日本分光製 Ubest-V570 紫外可視吸収分光光度計を使用し、測定セルは光路
長 1 cm の石英セルを使用した。サンプルは濃度を 0.25 ~ 1 mM の範囲で調製した錯体溶液
を用い、波長領域 900 ~ 300 nm の範囲において測定を行った。また、極低温での測定につ
いては UNISOKU の低温測定装置を分光器に取り付け、温度制御を行った。
128
6.2.5.2
電子スピン共鳴(ESR)スペクトル
測定装置は、JEOL JES-RE 1X ESR spectrometer を使用した。サンプルは濃度 1mM に調製
した錯体溶液を市販の ESR サンプルチューブに充填し、液体窒素を満たした測定用デュワ
ーに挿入し凍結させたのち、デュワーごと共振器に取り付けて測定を行った。測定条件を
以下に示した。Field, 3200±1000 G; Power, 1 mW; Sweep Time, 4 min; Modulation, 0.63 GHz;
Time Constant, 0.03 sec.
6.2.5.3
有機微量元素分析
測定装置は、Perkin Elmer 社製 2400II CHNS/O を使用した。試料測定前にガスブランク測
定を行った後、スズカプセルに封入した試料 1.5 ~ 2.0 mg を 2 回測定し、それを元素分析用
アセトアニリド標準試料による補正を行うことで C,H,N の各元素含有量(%)を求めた。
6.2.5.4
核磁気共鳴(1H-NMR)スペクトル
測定装置は、Varian Gemini 200 XL-300 型フーリエ変換核磁気共鳴装置を使用した。ケミ
カルシフトの基準物質として、テトラメチルシラン(TMS)を用いた。内径 5mmφ のサンプル
チューブ内に濃度を約 10 mM に調製した試料溶液について、積算回数を 16 に設定して δ=
-0.2 ~ 9.8 ppm の領域で測定した。
6.2.5.5
X 線結晶構造解析
回折データの測定には一辺が 0.1 ~ 0.3 mm の大きさの単結晶を用い、ガラスファイバー状
に結晶をグリースで固定し、-100 °C で測定した。格子定数は、6° < 2θ < 55°の範囲内の適当
な強度の回折点を用いて、最小二乗法により精密化を行った。
強度測定にはリガク社製 CCD 単結晶自動 X 線構造解析装置を用い、グラファイトで単色
化した Mo Kα線を X 線源とし、50 kV, 200 mA により測定した。強度が減衰する場合におい
ては decay correction による強度補正を行った。全反射データに対し Lorentz 因子及び偏光因
子の補正を加えたのち、I ≥ 2.00σ(I)の独立した反射を用いて解析を行った。
構造は重原子法により解析し、差フーリエ合成で得られなかった水素原子の座標は、結
晶水以外のものについては計算から求めた。非水素原子には異方性温度因子を適用し、更
に異常分散による補正、及び吸収補正を実行し、完全マトリックス最小二乗法で精密化を
行った。最小にした関数は、Σw(|F0|–|Fc|)2, w–1 = σ2(F0)である。原子散乱因子は International
Tables for Crystallography Vol.10を参照した。
構造解析、精密化は Crystal Structure 構造解析プログラム11により行い、計算は Windows
2000 オペレーティングシステムを搭載する市販のパーソナルコンピューターにて行った。
129
6.2.5.6
共鳴ラマン(rR)スペクトル
測 定 装 置 は 、 Ritsu Oyo Kogaku 社 製 Model MC-100DG spectrophotometer 、 Princeton
Instruments 社製 Model LN/CCD-1100-PB (Charge Coupled Device detector)を使用した。光源は、
Ar+イオンレーザーを用いた。測定は励起波長を 514.5 nm、サンプル濃度を 10 mM に調製し
た錯体溶液を高速回転用セルに充填し、低温条件下(-10 ~ -80 °C)にて測定を行った。
6.2.5.7
ESI-mass スペクトル
測定装置は、Micromass 社製 LCT (ESI-TOF 型)質量分析装置を使用した。錯体の濃度は約
50 µM に調製し、マイクロシリンジを用いて毎秒 600 µl/h の速度で溶液をシリンジポンプに
よって噴霧した。校正は NaI を用いて行い、データは Mass Lynx Ver. 3.5 を用いて Windows NT
ワークステーション上にて処理した。
6.2.5.8
ガスクロマトグラフ(GC)測定
測定装置として島津製作所製の GC-8APF を用い、検出器は FID、キャリアーガスには窒
素を用いた。解析は島津製作所製のクロマトパック C-R6A 及びクロマロパック C-R8A を
用い、最小ピーク幅を 5 秒、最小ピーク面積を 100 カウントとし、自動処置によりピーク
の面積を計算した。生成物の定量は内部標準法を用い、予め既知の量の反応生成物と内部
標準物質を用いて測定を行い、その面積比から検量線を作成し、その検量線に従って定量
した。以下にそれぞれの基質を用いた場合における分析条件を挙げる。
・チオアニソール
カラム:PEG-20M,
Chromosorb WAW DMCS、カラム長:3 m、カラム温度:205 ℃、イ
ンジェクション温度:240 ℃、キャリアーガス一次圧:600 kPa、キャリアーガス二次圧:
230 kPa、水素ガス圧:70 kPa、圧縮空気圧:60 kPa、内部標準物質:1-クロロナフタレン、
(保持時間)チオアニソール:2.2 min、フェニルメチルスルフォキシド:12.7 min、ジメチル
スルフォン:24.5 min.
・シクロヘキセン
カラム:PEG-20M,
Chromosorb WAW DMCS、カラム長:3 m、カラム温度:110 ℃、イ
ンジェクション温度:220 ℃、キャリアーガス一次圧:600 kPa、キャリアーガス二次圧:
180 kPa、水素ガス圧:70 kPa、圧縮空気圧:60 kPa、内部標準物質:1,2-ジクロロベンゼン、
(保持時間)シクロヘキセン:1.0 min、シクロヘキセンオキシド:3.7 min、シクロヘキセノン:
13.0 min、シクロヘキセノール:14.4 min
130
6.3
6.3.1
結果
FeII-PPBA 錯体の合成と構造
Fe-PPBA 錯体はカウンターアニオンの異なる二種類の結晶を得ることに成功し、
[FeII(ppba)(MeCN)2](ClO4)2 錯体は X 線による結晶構造解析に成功した。結晶学的パラメータ
を Table 6-1 に、主な結合長と結合角を Table 6-2 に、結晶の ORTEP 図を Figure 6-1 に示した。
錯体は鉄(II)イオンに対する 6 配位構造であり、溶媒であるアセトニトリル分子がシス位に
二分子配位していた。配位結合長は Fe-N(pyridine)が平均 2.163(9)Å、 Fe-N(amine)が 2.296(4)
Å、Fe-N(MeCN)が平均 2.159(0)Åであり、high-spin 型の鉄(II)錯体に特徴的な値であった。
[FeII(ppba)](OTf)2 錯体は、その層状構造のため X 線による構造同定ができなかったが、元素
分析より錯体はその空配位座にアセトニトリル分子が配位していないことが明らかとなっ
た。しかし、鉄(II)錯体は多座配位を好むため、空配位座には配位性のカウンターアニオン
であるトリフレートが配位して結晶化していると推定される。[FeII(ppba)](OTf)2 錯体のアセ
トニトリル溶液中における ESI-mass スペクトルを測定したところ、m/z = 205.1, 225.6, 246.1
に主なピークを観測した(Figure 6-2)。これらはそれぞれ[FeII(ppba)]2+, [FeII(ppba)(MeCN)]2+,
[FeII(ppba)(MeCN)2]2+錯体と同位体パターンが一致した。従って、アセトニトリル溶媒中で
は[FeII(ppba)](OTf)2 錯体も[FeII(ppba)(MeCN)2](ClO4)2 錯体と同様、空配位座に配位性の溶媒
分子であるアセトニトリルが配位していることが判明した。
6.3.2
FeII-PPBA 錯体と過酸化水素との反応
濃度 0.5 mM に調製した[FeII(ppba)](OTf)2 錯体のアセトン溶液を-78 oC に冷却し、10 当量
の過酸化水素を添加したところ、溶液は淡黄色から赤紫色に変化し、UV-vis スペクトルに
おいてλmax = 528 nm (ε = 1120 M-1cm-1)に特徴的な極大吸収を観測した(Figure 6-3)。この吸収
帯は過去に報告されているη1 型の単核鉄(III)-ハイドロパーオキソ錯体に見られるスペクト
ル的特徴に類似していることより、ハイドロパーオキソイオンから鉄(III)イオンに対する
LMCT と帰属される{[FeIII(tpa)(OOH)]2+ : 538 nm (ε = 1050 M-1cm-1), [FeIII(N4Py)(OOH)]2+ : 547
nm (ε = 1300 M-1cm-1), [FeIII(H2bppa)(OOH)]2+ : 568 nm (ε = 1200 M-1cm-1)}。この錯体溶液の
ESR スペクトルを測定したところ、high-spin 型の単核鉄(III)錯体に特徴的なスペクトルを示
し、算出された g 値は g// = 4.22, g⊥= 7.7 であった。同条件において調製した錯体溶液の共鳴
ラマンスペクトル測定の結果を Figure 6-4 に示した。H216O2 を用いて調製した錯体溶液を測
定したところ、824 cm-1, 842 cm-1 にパーオキソ種に由来すると考えられる O-O 間伸縮振動
のピークを観測した。また、H218O2 を用いて錯体溶液を調製したところ、880 cm-1 にラマン
シグナルを観測した。このことから、生成したハイドロパーオキソ錯体の酸素分子は添加
131
した過酸化水素に由来していることが明らかとなった。また、これらのラマンシグナルは
hign-spin 型の FeIII-OOH 錯体が示す O-O 間伸縮振動に特徴的な領域に観測された。
6.3.3
過酸化水素を酸化剤とした触媒反応
過去の研究より、合成した鉄(III)-ハイドロパーオキソ錯体[FeIII(ppba)(OOH)]2+は各種基質
に対する反応性を有することが予想される。そこで [FeII(ppba)]2+錯体を触媒とし、過酸化水
素を酸化剤とした基質の触媒的酸化反応を試みた。
反応は、錯体:基質 = 1:500 の割合で調製したアセトニトリル溶液 2 ml を、Ar 雰囲気
下において 0 oC に冷却しながら錯体に対し 100 当量の過酸化水素を添加することにより行
った。過酸化水素の添加は 15 分おきに 4 回行い、計 400 当量の酸化剤を添加した。また、
酸化反応生成物の定量は 15 分おきに GC 測定により行い、酸化反応の経時変化を追跡した。
反応はチオアニソールならびにシクロヘキセンを基質として行った(Scheme 6-1)。チオアニ
ソールを基質とした反応では触媒的な酸化反応の進行を確認し、GC 測定による酸化反応生
成物の定量結果を Table 6-3 に示した。反応の主生成物としてフェニルメチルスルフォキシ
ドが生成し、副生成物としてフェニルメチルスルホンの生成を確認した。また、過酸化水
素を逐次的に添加することによる触媒反応の継続を確認したが、その反応性は時間の経過
と共に減少した。シクロヘキセンを基質とした反応を行った場合も触媒的な酸化反応の進
行を確認し、GC 測定による酸化反応生成物の定量結果を Table 6-3 に示した。反応の主生成
物としてシクロヘキセノールが生成し、副生成物としてシクロヘキセノン、さらにはシク
ロヘキセンオキシドの生成を確認した。また、過酸化水素を逐次的に添加することによる
反応の継続を確認したが、チオアニソールの場合と同様、その反応性は時間の経過と共に
減少した。
132
6.4
考察
6.4.1
単核鉄(III)-ハイドロパーオキソ錯体の合成と性質
II
[Fe (ppba)]2+錯体と過酸化水素を反応させたところ、UV-vis スペクトルにおいてλmax = 528
nm (ε = 1120 M-1cm-1)に特徴的な極大吸収を観測した。この吸収帯は過去に報告されている
η1 型の単核鉄(III)-ハイドロパーオキソ錯体(FeIII-OOH)のものと非常に良い一致を示したこ
とから、ハイドロパーオキソイオンから鉄(III)イオンに対する LMCT と帰属される。ESR
スペクトルにおいて high-spin 型の単核鉄(III)種に特徴的なスペクトルを確認したことや、共
鳴ラマンスペクトルにおいても high-spin 型の FeIII-OOH 錯体に特徴的な領域にその O-O 間
伸縮振動が観測された。従って溶液中において単核の鉄(III)-ハイドロパーオキソ錯体が生
成していると考えられる。配位子場理論より Fe(III)イオンは 6 座の配位構造を好むことが知
られており、出発の[FeII(ppba)(MeCN)2](ClO4)2 錯体はシス位に二つの溶媒分子を伴った 6 配
位型の結晶であった。従って、[FeII(ppba)]2+錯体と過酸化水素を反応により生成する単核鉄
(III)-ハイドロパーオキソ錯体にはいくつかの構造異性体が考えられる。UV-vis スペクトル
の特徴から、錯体は end-on 型の FeIII-OOH 錯体である可能性が高い。錯体はシス位に二つの
空配位座を有しているため、その構造は 5 配位型錯体と溶媒等を伴った 6 配位型錯体が考
えられる。H216O2 を用いた共鳴ラマンスペクトル測定においては、16O-16O 間伸縮振動に対
応すると考えられるピークが 824 cm-1, 842 cm-1 に 2 つ観測されたため、測定条件によりこの
ような構造異性体が存在している可能性が示唆された。
6.4.2 [FeIII(ppba)(OOH)]2+錯体の酸化反応特性
過酸化水素を酸化剤とし、[FeII(ppba)]2+錯体を触媒としたチオアニソール、シクロヘキセ
ンの酸化反応を行ったところ、それぞれ触媒的な酸化反応の進行を確認した。低温条件下
における[FeII(ppba)]2+錯体と過酸化水素の反応では、[FeII(ppba)(OOH)]2+錯体の生成を確認し
ていることから、反応系中においてはこのような化学種を中間体とした反応が進行してい
ると考えられる。チオアニソールを基質とした反応を行ったところ、フェニルメチルスル
フォキシドとフェニルメチルスルホンが酸化生成物として生成した。フェニルメチルスル
ホンは酸化生成物であるフェニルメチルスルフォキシドの再酸化により生成していると考
えられる。従って、活性種はスルホンに対して高い選択性を有していることが明らかとな
った。これらの基質は硫黄原子の電子密度よりスルフィドは求電子的な活性種に対して高
い反応性を示し、スルフォキシドはスルフィドに比べ求核的な活性種により高い反応性を
示すことが指摘されている(Scheme 6-2)。従って、[FeII(ppba)(OOH)]2+錯体、もしくはその分
解により生成する反応活性種は求核的な反応性も有していることが判明した。鉄(III)-ハイ
133
ドロパーオキソ錯体が示すこのような求核的な反応性は 4, 5 章で報告した CuII-OOH 錯体に
は見られなかった。また、過去に合成された[FeIII(H2bppa)(OOH)]2+錯体は high-spin 型の鉄
(III)-ハイドロパーオキソ錯体であり、有機硫黄化合物に対する求核的な反応性を示すこと
が報告されている 7。シクロヘキセンを基質とした反応を行ったところ、酸化生成物として
シクロヘキセンオキシド、シクロヘキセノール、シクロヘキセノンの生成を確認した。反
応ではシクロヘキセノールが最も選択的に生成したが、4, 5 章で報告した CuII-OOH 錯体に
は見られなかったシクロヘキセンオキシドの生成も確認した。シクロヘキセンオキシドは
オレフィンの求電子的な酸素原子の付加反応で生成することが知られている。その収率は
低いものの、[FeII(ppba)(OOH)]2+錯体はこのような反応性も有していることが明らかとなっ
た。また、オレフィンに対するその酸化反応活性は 4, 5 章で報告した CuII-OOH 錯体よりも
高いことが判明した。
134
6.5
結論
より活性の高い錯体触媒の開発を目的とし、中心金属に鉄(II)イオンを有する単核錯体を
合成し、過酸化水素との反応により単核鉄(III)-ハイドロパーオキソ錯体の合成を行った。
また、触媒条件下での酸化反応実験をおこない、単核鉄(III)-ハイドロパーオキソ錯体が示
す反応性を検討した。
合成した鉄(II)錯体は、四座配位子 PPBA を用いることにより、ハイドロパーオキソイオ
ンの活性化に必要と考えられているシス位の空配位座を有していた。従って、合成した単
核鉄(III)-ハイドロパーオキソ錯体にはいくつかの構造異性体が存在すると考えられるが、
そのスペクトル的特徴より end-on 型の単核錯体と推定される。また、合成した単核鉄(III)ハイドロパーオキソ錯体は high-spin 型であることが判明した。このような high-spin 型の単
核鉄(III)-ハイドロパーオキソ錯体は基質に対し求核的な反応性を有することが報告されて
いるが
7,12
、現在その詳細な反応機構等は解明されていない。[FeIII(ppba)(OOH)]2+ 錯体は
high-spin 型であり、且つ錯体に配位しているハイドロパーオキソイオンのシス位に空配位
座を持つ単核鉄(III)-ハイドロパーオキソ錯体であり興味深い。触媒条件下においてチオエ
ーテルを基質とした反応を行ったところ、酸化生成物としてスルフォキシドとスルホンが
生成した。スルホンの生成はスルフォキシドの再酸化により生成すると考えられるため、
錯体は求核的な反応性を有することが判明した。このような求核的な反応性は銅錯体を用
いた単核銅(II)-ハイドロパーオキソ錯体では見られず、過去に Wada らが報告している
[FeIII(H2bppa)(OOH)]2+錯体の示す反応性に類似していた。シクロヘキセンの酸化においては、
シクロヘキセノールの収率が最も高く選択的な酸化反応が観測されたが、シクロヘキセン
オキシドとシクロヘキセノンの生成も確認した。求電子的な酸素分子の挿入反応によるシ
クロヘキセンオキシドの生成は単核銅(II)-ハイドロパーオキソ錯体では見られない反応特
性であった。
中心金属に鉄を有する単核鉄(III)-ハイドロパーオキソ錯体を合成したところ、チオエー
テル類の酸化反応に対しては求核的な反応特性を示し、またオレフィンの酸化に対しては
前章までに報告した単核銅(II)-ハイドロパーオキソ錯体よりも高い反応性を有することが
明らかとなった。合成した[FeIII(ppba)(OOH)]2+錯体はシス位に空配位座を持つ high-spin 型の
単核鉄(III)-ハイドロパーオキソ錯体である。このような錯体を用いた酸化反応特性に対す
る報告は非常に少なく 7、その酸化反応パラメータを収集することは反応機構の解明のため
非常に重要な知見と考えられる。
135
References
1
(a) Ford-Hutchinson, A. W.; Gresser, M.; Young, R. N. Annu. Rev. Biochem. 1994, 63, 383.
(b) Kühn, H.; Borngräber, S. In Lipoxygenases and Their Metabolites, Nigam, S.; Pace-Asciak,
C. R. eds., Plenum, New York, 1999.
2
(a) Ramaswamy, S. In Handbook on METALLOPROTEINS, Bertini, I.; Sigel, A.; Sigel, H. eds.,
Marcel Dekker, Inc., New York, 2001, pp 613. (b) Bertini, I.; Cermonini, M. A.; Ferretti, S.;
Lozzi, I.; Luchinat, C.; Viezzoli, M. S. Coord. Chem. Rev. 1996, 151, 145.
(c) Karlsson, A.;
Parales, J. V.; Parales, R. E.; Gibson, D. T.; Eklund, H.; Ramaswamy, S. Science 2003, 299,
1039.
3
(a) Solomon, E. I.; Brunold, T. C.; Davis, M. I.; Kemsley, J. N.; Lee, S. -K.; Lehnert, N.; Neese,
F.; Skulan, A. J.; Yang, Y. -S.; Zhou, J. Chem. Rev. 2000, 100, 235.
(b) Girerd, J. -J.; Banse, F.;
Simaan, A. J. in Structure and Bonding , Meunier, B. ed., Springer, Verlag, Berlin, 2000, 97, pp
145.
4
(a) Umezawa, T. Gann Monogr. Cancer Res. 1976, 19, 3.
(b) Stubbe, J; Kozarich, J. W.; Wu,
W.; Vanderwall, D. E. Acc. Chem. Res. 1996, 29, 322. (c) Burger, R. M. Chem. Rev. 1998, 98,
1153. (h) Claussen, C. A.; Long, E. C. Chem. Rev. 1999, 99, 2797.
5
(a) Kim, J.; Harrison, G.; Kim, C.; Que, L., Jr. J. Am. Chem. Soc. 1996, 118, 4373. (b) Kim, C.;
Chen, K.; Kim, J.; Que, L., Jr. J. Am. Chem. Soc. 1997, 119, 5964.
G.; Feringa, B.; Que, L., Jr. J. Am. Chem. Soc. 1999, 121, 264.
(c) Ho, R. Y. N.; Roelfes,
(d) Kim, J.; Larka, E.;
Wilkinson, E. C.; Que, L., Jr. Angew. Chem. Int. Ed. Engl. 1995, 34, 2048.
(e) Kim, J.; Dong,
Y.; Larka, E.; Que, L., Jr. Inorg. Chem. 1996, 35, 2369. (f) Zang, Y.; Kim, J.; Dong, Y.;
Wilkinson, E. C.; Appelman, E. H.; Que, L., Jr. J. Am. Chem. Soc. 1997, 119, 4197.
H.; Chen, K.; Lange, S. J.; Que, L., Jr. Inorg. Chem. 2001, 40, 3534.
(g) Miyake,
(h) Zang, Y.; Elgren, T.
E.; Dong, Y.; Que, L., Jr. J. Am. Chem. Soc. 1993, 115, 811.
6 (a) Chen, K.; Que, L., Jr. Angew. Chem. Int. Ed. Engl. 1999, 38, 2227. (b) Chen, K.; Que, L., Jr.
J. Am. Chem. Soc. 2001, 123, 6327.
(c) Chen, K.; Costas, M.; Kim, J.; Tipton, A. K.; Que, L.,
Jr. J. Am. Chem. Soc. 2002, 124, 3026. (d) Chen, K.; Costas, M.; Que, L., Jr. J. Chem. Soc.
Dalton Trans. 2002, 672. (e) Fujita, M.; Costas, M.; Que, L., Jr. J. Am. Chem. Soc. 2003, 125,
9912.
7
Wada, A.; Ogo, S.; Nagatomo, S.; Kitagawa, T.; Watanabe, Y.; Jitsukawa, K.; Masuda, H. Inorg.
Chem. 2002, 41, 616.
8
S. Yamaguchi, Ph.D Dissertation of Nagoya Institute of Technology, 2004
9
M. Harata, K. Jitsukawa, H. Masuda and H. Einaga. Chem. Lett., 1995, 61-62.
10 International Tables for X-ray Crystallography, Ibers, J. A.; Hamilton, W. C. eds., Kynoch Press,
Birmingham, U. K., 1974, Vol. IV.
11 “Crystal Structure” analysis program (ver 3.7) Produced by Rigaku, 2005
12 M. Costas, M. P. Mehn, M. P. Jensen and L. Que. Jr., Chem. Rev., 2004, 104, 939-986.
136
O
N
N
H
N
N
PPBA
Chart 6-1 Ligand used in this work
Table 6-1 Crystallographic data and refinement parameters for [FeII(ppba)(MeCN)2](ClO4)2.
[Fe(ppba)(CH3CN)2](ClO4)2
C25H36Cl2FeO9N6
691.35
Yellow,block
0.20×0.20×0.20
monoclinic
P 21/c (#14)
9.828(6)
30.18(2)
11.191(7)
103.439(7)
3228.9(35)
4
1.422
1440.00
MoKα(λ=0.71070Å)
Rigaku/MSC mercury CCD
-100.0
55.0
Total:25752
4900
424
11.56
0.069/0.135
1.24
0.03
Complex
Empieical Formula
Formula Weight
Crystal Color
Crystal Dimensions / mm
Crystal System
Space Group
a/ Å
b/ Å
c/ Å
β/ deg
Cell Volume / Å3
Z value
-3
Dcalc / gcm
F(000)
Radiation
Detectometer
T/℃
2θ max /deg
No.of Reflections Measured
No.of Observations
No.Variables
Reflection/Parameter Ratio
R / Rw
G.O.F
Max Shift / Error
137
Table 6-2 Selected bond lengths (Å) and angles (o) of [FeII(ppba)(MeCN)2](ClO4)2.
Fe(1)-O(1)
Fe(1)-N(1)
Fe(1)-N(2)
O(1)-Fe(1)-N(1)
N(1)-Fe(1)-N(2)
N(1)-Fe(1)-N(4)
O(1)-Fe(1)-N(5)
N(2)-Fe(1)-N(5)
O(1)-Fe(1)-N(6)
N(2)-Fe(1)-N(6)
N(5)-Fe(1)-N(6)
Bond Length (Å)
2.039(3)
Fe(1)-N(4)
2.296(4)
Fe(1)-N(5)
2.175(4)
Fe(1)-N(6)
Bond Angles (deg)
156.3(1)
O(1)-Fe(1)-N(2)
76.7(2)
O(1)-Fe(1)-N(4)
77.9(2)
N(2)-Fe(1)-N(4)
95.2(2)
N(1)-Fe(1)-N(5)
172.4(2)
N(4)-Fe(1)-N(5)
96.6(2)
N(1)-Fe(1)-N(6)
86.2(2)
N(4)-Fe(1)-N(6)
86.6(2)
2.152(4)
2.170(5)
2.147(4)
83.4(2)
92.8(2)
98.5(2)
106.2(2)
89.0(2)
94.7(2)
170.0(2)
Figure 6-1 ORTEP drawing for cation part of [FeII(ppba)(MeCN)2](ClO4)2 with the atom
labeling scheme. The thermal ellipsoids are at the 50% probability level, and the hydrogen
atoms were omitted for clarity.
138
Figure 6-2 ESI-mass spectrum of [FeII(ppba)](OTf)2 complex in MeCN solution.
0.7
0.6
Absorbance
0.5
0.4
0.3
0.2
0.1
0
300
400
500 600 700
Wavelength [nm]
800
900
Figure 6-3 UV-vis spectral change of the reaction of [FeII(ppba)]2+ with H2O2 at -78 oC in
acetone. (a) [FeII(ppba)](OTf)2 (0.5 mM) (b) add H2O2 10eq
139
824 cm-1 842 cm-1
880 cm-1
760
780
800
820
840
860
880
900
Figure 6-4 Resonance Raman spectra for the reaction product of [FeII(ppba)]2+ with H216O2
(top) and H218O2 (bottom) in acetone at -80 oC.
O
S
O
S
FeIIPPBA, H2O2
S
+
MeCN, 0°C, Ar
Phenylmethylsulfoxide
Thioanisole
O
Phenylmethylsulfone
OH
FeIIPPBA, H2O2
O
MeCN, 0°C, Ar
Cyclohexene
Cyclohexene oxide
+
O
+
Cyclohexenol
Scheme 6-1 Substrates and their oxidation products in the reaction.
140
Cyclohexenone
Table 6-3 Summary of the catalytic oxidation by the FeII-PPBA complex with hydrogen
peroxide. (TON exhibits a “turn over number”)
time
[min]
15
30
45
60
H2O2
[eq]
100
+100
+100
+100
Thioanisole [TON]
Sulfoxide
Sulfone
56
7
85
17
95
20
102
21
Cyclohexene [TON]
Epoxide
Alcohol
Ketone
2.2
7.6
4.4
3.5
13.1
7.4
4.6
17.3
8.5
4.8
19.1
9.3
δ-
O
δ-
S
S
δ+
electrophilic
or
radicalitic attack
nucleophilic attack
Scheme 6-2 Difference of the reactivity of organic sulfur substrates.
141
142
第7章
総括
本研究では銅含有酸化酵素である DβM, PHM の活性中間体を、錯体分子を用いて合
成し、その物性解析と反応性の検討を行った。これら酵素の反応中間体として現在主に
単核銅(II)-スーパーオキソ種を活性種とする機構と、単核銅(II)-ハイドロパーオキソ種
を活性種とする機構が提案されている。そこで、研究はこれらの反応機構に対応させる
べく 2 つの戦略に基づいて行った。ひとつは「単核銅(II)-スーパーオキソ錯体の合成と
その反応性の検討」であり、 ひとつは「単核銅(II)-ハイドロパーオキソ錯体の合成と
その反応性の検討」である。これらの結果を比較することより、提案されている酵素の
反応機構を実験化学的に評価した。また、さらに活性の高い錯体触媒の開発を目指し中
心金属に鉄を用いた錯体の合成を行い、その酸化反応特性を評価した。
第 1 章では、生体内に存在する酵素とその活性中心に存在する金属イオンの役割につ
いて述べた。研究では特に銅含有酸化酵素である DβM, PHM に着目し、過去に合成さ
れたこれら酵素のモデル錯体をまとめ、その問題点について言及した。その上で、生物
無機化学的手法により酵素の反応機構を明らかにするために必要な研究の戦略を述べ
た。
第 2 章では、[CuI(H2bppa)]+錯体と酸素分子を反応させ、単核銅(II)-スーパーオキソ錯
体を経由した単核銅(II)-ハイドロパーオキソ錯体の合成を行った。このような反応は
DβM, PHM の活性中心で進行していると考えられているが、それをモデル錯体により再
現した例はない。実験では酵素の反応機構に基づいた単核銅(II)-ハイドロパーオキソ錯
体の合成に成功しており、酵素内部でこのような反応が進行し得ることを実験化学的に
明らかにした。
第 3 章では、[CuI(bnpa)]+錯体を用いて合成した単核銅(II)-スーパーオキソ錯体の外部
基質に対する水素原子引き抜き反応を行った。単核銅(II)-スーパーオキソ種は、DβM,
PHM において推定されている反応中間体の一つであるが、実験化学的にその反応性を
確認した例はない。合成した単核銅(II)-スーパーオキソ種は BDE < 72.6 kcal mol-1 の基質
に対する反応性を有することが明らかとなった。
第 4 章では、配位子 BPBA を用いた単核の銅(II)-ハイドロパーオキソ錯体の合成を行
った。過去に合成された単核銅(II)-ハイドロパーオキソ錯体の多くは 5 配位型錯体であ
るのに対し、[CuII(bpba)(OOH)]+錯体は歪んだ平面構造を有する 4 配位型錯体であった。
[CuII(bpba)(OOH)]+錯体は過去に合成された錯体群と比べ、その生成ならびに分解速度が
大きく増加していた。また、[CuII(bpba)(OOH)]+錯体は 5 配位型錯体に比べ高い反応性を
143
示し、チオエーテルやアリル化合物、芳香環α位に対する反応性を有することが明らか
となった。
第 5 章では、配位子に BPBA, BPIPA, BPEA を用いた単核銅(II)-ハイドロパーオキソ錯
体の比較を行った。これらの錯体は配位子のアルキル置換基の立体障害により 4 配位型
錯体間で異なった平面構造を有しており、錯体の平面性は BPBA 錯体 < BPIPA 錯体 <
BPEA 錯体の順に増加していた。単核銅(II)-ハイドロパーオキソ錯体の生成・分解速度
ならびに基質に対する反応性は 3 錯体で大きく異なり、4 配位型錯体間でも構造の僅か
な違いが錯体の物性に大きな影響を及ぼすことが明らかとなった。
第 6 章では、より高い触媒活性の発現を目的とし、単核鉄(III)-ハイドロパーオキソ錯体
を用いた各種基質の酸化反応を行った。錯体は 4 座型配位子 PPBA を用い、シス位に空配
位座を有する high-spin 型錯体であることが判明した。錯体は有機硫黄を基質とした場合、
スルフォキシドに対する求核的な反応性を有し、シクロヘキセンの反応においては前章で
報告した単核銅(II)-ハイドロパーオキソ錯体より高い反応性を有することが明らかとなっ
た。
研究では、推定されている 2 種類の反応中間体「単核銅(II)-スーパーオキソ種」と「単
核銅(II)-ハイドロパーオキソ種」をモデル錯体により合成し、外部基質に対する反応性
を評価することに成功した。結果、本錯体系においては単核銅(II)-ハイドロパーオキソ
錯体は単核銅(II)-スーパーオキソ錯体に比べ、基質に対しより高い反応性を示した。合
成した反応中間体の基本的物性や反応性は、その配位構造の僅かな差異に著しく影響さ
れることが明らかとなり、研究では錯体分子の厳密な設計を行うことによりその反応性
の制御に成功したと言える。このように厳密な分子設計に基づく活性中間体の反応制御
は、酵素の高い活性機構を応用した機能性分子の創成につながる重要な知見と考えられる。
物質の酸化反応は産業的な観点からも非常に重要であり、将来的には一般的に高い反応
性を示す希少金属に代わり自然界に豊富に存在する安価な金属を用いた高機能触媒が用い
られるべきである。本研究では、そのような機能性分子の創成における生物無機化学的な
手法の有効性を、銅含有酸化酵素のモデル化学的な研究により確認した。これをもって本
論文の総括とする。
144
発表論文
-第 2 章Tatsuya Fujii, Syuhei Yamaguchi, Yasuhiro Funahashi, Tomohiro Ozawa, Takehiko Tosha, Teizo
Kitagawa and Hideki Masuda
“Mononuclear copper(II)-hydroperoxo complex derived from reaction of copper(I) complex with
dioxygen as a model of DβM and PHM”, Chem. Commun., 4428-4430 (2006)
-第 3 章Tatsuya Fujii, Syuhei Yamaguchi, Koichiro Jitsukawa and Hideki Masuda
“H-atom abstraction reaction for organic substrates via mononuclear copper(II)-superoxo species as
a model of DβM and PHM ”, Dalton Trans., in submission
-第 4 章Tatsuya Fujii, Asako Naito, Syuhei Yamaguchi, Akira Wada, Yasuhiro Funahashi, Koichiro
Jitsukawa, Shigenori Nagatomo, Teizo Kitagawa and Hideki Masuda
“Construction of a square-planar hydroperoxo-copper(II) complex inducing a higher catalytic
reactivity”, Chem. Commun., 2700-2701 (2003)
-第 5 章Tatsuya Fujii, Asako Naito, Koichiro Jitsukawa and Hideki Masuda
“Synthesis and characterization of mononuclear copper(II)-hydroperoxo complexes with tridentate
pyridylmethyl ligands; Reaction of exogenous substrates via copper(II)-hydroperoxo species”, in
preparation.
-第 6 章Tatsuya Fujii, Tomohiro Ozawa, Yasuhiro Funahashi, Koichiro Jitsukawa and Hideki Masuda
“Synthesis of Mononuclear Non-Heme Iron(III)-Hydroperoxo Complex as an Oxidative Catalyst”,
Advanced Materials Research Vols. 11-12, 331-334 (2006)
145
参考論文
1. Tatsuya Fujii, Asako Naito, Syuhei Yamaguchi, Akira Wada, Yasuhiro Funahashi, Koichiro
Jitsukawa and Hideki Masuda, “Oxidation Activity of Hydroperoxo-copper(II) Complex as a
DβH”, J. Inorg. Biochem., 96, 134 (2003).
2. Takeshi Kato, Tatsuya Fujii, Tomohiro Ozawa, Yasuhiro Funahashi and Hideki Masuda
“[N,N-Bis(carboxymethyl)-L-leucinato]-(1,2-ethanediamine)cobalt(III)
monohydrate”,
Acta
Cryst., E62, m92-m94.
3. 増田秀樹、大畑奈弓、奥村健志、藤井達也
「環境対応型セラミックスの技術と応用」 第 3 編「環境・エネルギーへの応用技術」
第 7 章「触媒技術 ~分子レベルでの精密設計からナノ反応場開発戦略まで~」
,CMC 出版, 274-292 (2007)
146
謝辞
本研究は名古屋工業大学大学院工学研究科物質工学専攻増田研究室において、増田秀樹
教授の適切なる御指導のもと、大学院工学研究科博士後期課程の研究として行ったもので
あります。ここに、増田秀樹教授に対し深く感謝の意を表するとともに謹んで御礼申し上
げます。また、研究のみならず、化学に対する姿勢についても御教示して頂いた実川浩一
郎教授に厚く御礼申し上げます。
研究を行うにあたり、日頃より叱咤・激励して頂いた小澤智宏助教授、実験に関して御
助言を頂きました舩橋靖博博士に厚く御礼申し上げます。また、研究室の円滑な運営に御
尽力下さいました技術課職員、谷山八千代さんに御礼申し上げます。
共鳴ラマンスペクトルの測定においてお世話になりました自然科学研究機構統合バイオ
サイエンスセンターの北川禎三教授、長友重紀博士、当舎武彦博士に厚く御礼申し上げま
す。また、博士後期課程在学中、研究費等を御支援下さいました名古屋工業大学 21 世紀 COE
プログラムに厚く御礼申し上げます。
日頃より的確な御助言をして頂いた山口修平博士、有井秀和博士、松本健司博士、猪股
智彦博士、梶田裕二博士、加藤貴志博士、大畑奈弓博士を始めとする諸先輩方に深く感謝
致します。長きに渡る苦楽を共にした奥村健志氏、高橋勇雄氏を始めとする同期の皆様に
深く感謝致します。また、研究を始め日々の生活を共にした矢野卓真君、水谷洋介君、中
根大輔君を始めとする増田研究室の皆様に深く感謝致します。
最後に、長きに渡る学生生活を精神的、金銭的に支えて下さった両親、家族に深く感謝
の意を表し、これをもって謝辞とさせて頂きます。
2007 年 3 月
藤井
147
達也
© Copyright 2026 Paperzz