ISSN 1348 – 4052
東
北
大
學
多 元 物 質 科 学 研 究 所
素材工学研究彙報
第 59 巻 第 1,2 號
2003 年(平成 15 年)12 月
Bulletin
of the
Advanced Materials Processing Building, IMRAM
Tohoku University
(SOZAIKEN IHO)
Vol.59, No.1, 2 December 2003
Sendai, Japan
多元物質科学研究所
素材工学研究彙報
第 59 巻
目
次
報
文
非質量分離型イオンビームデポジションによる Ta 拡散バリア膜の作製と
評価·····················································································································
林
載 元
裵
俊 佑
········
三 村 耕 司
一 色
実
1
胴長の異なる転動ミル内における媒体運動と粉砕速度との相関··················
三 尾
浩
加 納 純 也 ········
齋 藤 文 良
10
集束イオンビーム法と電子線ホログラフィーを用いた Fe‑Co‑Cu‑Nb‑Si‑B
ナノ結晶磁性材料の磁区構造評価 ···································································
斉
朴
村
進
吉
17
光誘導ドリフトによる同位体分離 ···································································
武 山 昭 憲
佐 藤 俊 一
藤 健
英
上 恭
藤 大
沢 克
太
吉
和 ········
輔
仁
········
22
サラントヤ
ミャグマルジャブ
液相還元選択析出法による Ni‑Zn ナノ粒子の成長機構と物性······················
資
高
砂
山
佐
村
橋
川
本
藤
松
英
洋
勝
修
淳
志
二 ·········
俊
彰
司
28
料
希土類−シリコン−酸窒化物 RE 4Si2O7N2 (RE‑J‑phase, RE=希土類)の結晶構
造·························································································································
高 橋 純 一
山 根 久 典 ········· 3 6
島 田 昌 彦
CONTENTS
Original Papers
Fabrication and Characterization of Ta Diffusion Barriers Deposited by Non‑Mass Separated
Ion Beam Deposition ······································································································································
1
By Jae‑Won LIM, Joon Woo BAE, Kouji MIMURA and Minoru ISSHIKI
Correlation of Grinding Rate with Specific Ball Impact Energy Simulated in Tumbling Mills
with Different Pot Lengths·························································································································
10
By Hiroshi MIO, Junya KANO and Fumio SAITO
Analysis of Magnetic Domain Structure in Fe‑Co‑Cu‑Nb‑Si‑B Nanocrystalline Magnetic
Materials by Focused Ion Beam Method and Electron Holography ························································
By Kenta SAITO, Young‑Gil PARK, Yasukazu MURAKAMI, Daisuke SHINDO and
17
Yoshihito YOSHIZAWA
Isotope Enrichment of Lithium by Lignt‑induced Drift ·······································································
By Akinori TAKEYAMA and Shunichi SATO
22
Growth Mechanism and Characterization of the Ni‑Zn Nanoparticles on TiO2 Fine Particles
Synthesized by the Liquid Phase Reductive Deposition Method··························································
By Sarantuya MYAGMARJAV, Hideyuki TAKAHASHI, Yoji SUNAGAWA, Katsutoshi YAMAMOTO,
28
Nobuaki SATO and Atsushi MURAMATSU
Review Articles
Crystal Structure of Rare‑earth Silicon‑oxynitrides RE4Si2O7N2 (RE‑J‑phase, RE = Rare Earth)
··········································································································································································
By Junichi TAKAHASHI, Hisanori YAMANE and Masahiko SHIMADA
“SOZAIKEN” is the abbreviation of SOZAI KOUGAKU KENKYUUTOU
(Advanced Materials Processing Building)
SOZAI means ORDINARY and NEW MATERIALS and KEN means RESEARCH INSTITUTE.
36
非質量分離型イオンビームデポジションによる Ta 拡散バリア
膜の作製と評価
林
載元*, 裵
俊佑*, 三村
耕司*, 一色
実*
Fabrication and Characterization of Ta Diffusion Barriers Deposited by Non-Mass Separated
Ion Beam Deposition
By Jae-Won LIM, Joon Woo BAE, Kouji MIMURA and Minoru ISSHIKI
The interfacial reactions of the Cu(100nm)/Ta(50nm)/Si structures and their relationship with the
microstructure of Ta diffusion barriers are investigated. The Ta films were deposited on Si (100) substrates at
various bias voltages ranging from 0 to -200 V. The Ta diffusion barriers which were deposited at the substrate
bias voltages of -50 V and -125 V prevented Cu-Si interaction up to 600°C in flowing purified H2 for 60 min,
whereas the Ta layer with a columnar structure which was deposited at zero bias voltage degraded at 400°C. It
was found that a slight resistivity increase of the Cu/Ta(-50 V or -125 V)/Si structures at 650°C seemed to be
due to a Cu agglomeration. To confirm the thermal stability of the Ta diffusion barrier deposited at the substrate
bias voltage, a SiO2 capping layer was used as a suppressor and was deposited on the Cu/Ta(-125 V)/Si
structure. As a result, the Cu/Ta(-125 V)/Si structures could be stable up to 650°C without the Cu-Si interaction.
Two different reactions of the Cu/Ta(0 V)/Si and the Cu/Ta(-50 V or -125 V)/Si structures concerning the
thermal stability were discussed on the basis of the experimental results.
(Received October 3rd, 2003)
Keywords: tantalum, diffusion barrier, ion beam deposition, resistivity, substrate bias voltage, thermal stability
緒言
ULSI の超高集積化が進むにつれて,デバイス回路全体の動作速度は,能動素子のトランジスタの
性能よりむしろ,受動素子である配線の性能で律速されるようになる.これは,デバイスチップ上で
トランジスタは配線で相互に接続されており,R‑C(Resistance‑Capactance: 抵抗‑容量積)時定数で
充放電しながら信号を伝達していくため,配線幅,間隔の寸法を縮小すると配線抵抗と配線間容量が
増大し配線遅延が増大するからである[1,2].したがって,配線縮小に伴い,配線材料として Al(2.7
μΩcm)の代わりに比抵抗 1.67 μΩcm の Cu の導入が要求され,近年 Si デバイスに対する Cu 配線材
料の適用が実用化され始めている[3].
しかし,Cu 配線材料を Si デバイスに完全に適用するためには,
まだいくつかの課題を解決しなければならない.その中の一つは,Si 基板あるいは層間絶縁膜への Cu
原子の拡散によるトランジスタ特性の劣化である.このような Cu の Si 中への拡散を防ぐため,バリ
ア層の形成が必要である.
このバリア材料に対しては,(1)高融点の材料で高耐熱性を持つ,(2)銅との化合物を作らない,
(3)低抵抗である,(4)表面平坦性がよい,(5)密着性が高いことなどが要求される[4].以上のことを
考慮すると拡散防止膜としては高融点金属およびその窒化物(Ti, TiN, Ta, TaN, W, WN)が適している.
この中でタンタル(Ta)は高融点で低抵抗の薄膜が可能で,Cu 膜との密着性がよく,銅と反応せず比較
1.
* 東北大学多元物質科学研究所
林
載元, 裵
俊佑, 三村
耕司, 一色
第 59 巻 第 1,2 号
実
的安定な界面を形成すると報告されている[5].Table 1 に種々な方法で作製されたタンタル拡散防止
膜の耐熱安定性に関する報告結果をまとめて示した.Cu と Si の反応温度範囲は 300°C〜650°C でかな
り幅があることが分かる[6‑11].これは作製したバリア膜の微細構造,すなわち多結晶構造を有する
膜の主たる高速拡散経路である結晶粒界が原因と思われる.バリア材の拡散防止能を向上させるには,
粒界に沿った Cu 原子の拡散をいかに抑制するかが重要な鍵となる.一般のスパッタリングでは柱状結
晶を有する膜ができ,Cu 原子の粒界拡散により Si との反応温度が低くなると言われ,拡散防止能向
上の手法として柱状結晶を持たないより緻密な薄膜が要求される.
Table 1 Failure temperatures of Ta diffusion barrier in Cu/Ta/Si structures.
Sample structure
(nm thickness)
Cu(200)/Ta(100)/Si
Fabrication method
Annealing
time (min)
30
Failure
temperature (℃)
300-400
Ref.
Electron beam evaporation
Annealing
atmosphere
N2:H2(9:1)
Cu(200)/Ta(180)/Si
Sputtering
Vacuum(10-5 Pa)
30
550
7
Cu(100)/Ta(50)/Si
Magnetron sputtering
He
30
500-550
8
Cu(200)/Ta(100)/Si
sputtering
Vacuum(10-5 Pa)
60
600
9
Cu(100)/Ta(30)/Si
Electron beam evaporation
Vacuum(10-4 Pa)
30
550
10
Vacuum(10-4 Pa)
30
650
Cu(50)/Ta(Vb)(50)/Si
Electron beam evaporation +
ion beam assisted deposition
Optimum bias sputtering
N2
30
650
Cu(100)/Ta(unbiased)(50)/Si
Ion beam deposition
H2
60
400
Cu(100)Ta(biased)(50)/Si
Ion beam deposition
H2
60
650
6
11
This
work
薄膜作製技術として,真空蒸着,スパッタリング,CVD などの方法があるが,これらは,電気的
に中性状態にある金属粒子で成膜を行うため,基板表面に入射する堆積粒子のエネルギーを制御する
のは難しい.この問題に対して,エネルギーを制御できる質量分離型 IBD(イオンビームデポジション)
法が高純度物質や新機能性材料の作製方法の一つとして大きな関心を集め,研究が増加する傾向にあ
る[12‑14].質量分離型 IBD 法はイオンの運動エネルギーなど,成膜を支配する多くのパラメータを電
気的に制御でき,単に物質を分離して高純度化するだけでなく,成膜された薄膜に多くの機能性を与
えることができる.したがって,他の方法では高温においてしか形成できない物質,あるいは準安定
状態においてしか存在しない物質を,低い基板温度領域において形成することができる.ところで,
質量分離型 IBD 法は金属薄膜に関する基礎的研究やその解析に対して多く貢献しているが,蒸着され
る膜の面積が小さく(直径 10 mm 以内)
,低効率性,高価の装備など実用的応用には不向きである.こ
れに比べ,最近,質量分離を行わない安価な非質量分離型 IBD 装置が開発され,磁性材料,半導体材
料および銅配線材料への応用の道が開かれた[15,16].非質量分離型 IBD はシンプルな構造で広い蒸着
面積(直径 50mm 以上)を持ち,高効率の膜作製が可能である.また,RF スパッタ型イオン源の開発
により銅やタンタルなど大半の金属をソースに利用できる[17].
本研究では,高純度の銅あるいはタンタルを RF スパッタ型イオン源に用い,非質量分離型 IBD
法によるタンタルバリア拡散防止膜の作製方法および作製した膜の特性に関して検討した.
実験方法
非質量分離型 IBD 装置の概略を Fig. 1 に示す.チャンバー下部に取り付けた二つのイオン源は,
プラズマ励起用 RF コイル(内径 57.5 mm,巻数 5)内部に棒状の銅(6N,8 mm),タンタル(4N,10 mm)
2.
平成 15 年 12 月
非質量分離型イオンビームデポジションによる Ta 拡散バリア膜の作製と評価
Negative DC
Bias Voltage
(VS)
Doposition Chamber
Load-Lock Chamber
Substrate Holder
Substrate
Shutter
Cu Grid
Impedance
Matching
Box
Ar gas(PAr)
RF Power Supply
(13.56MHz)
Cu Coil
Impedance
Matching
Box
Cu Target
Ta Target
Turbomolecular pump
Negative DC
Bias Voltage
(VT)
Fig. 1 Schematic diagram of the non-mass separated ion
beam deposition apparatus.
を配置したものである.銅ターゲットは電
解研磨,タンタルターゲットは化学研磨に
より,表面汚染層を十分除去した.基板に
は,Si(100)を用い,アセトンで超音波洗浄
後,5%HF により表面酸化層を除去した.Si
基板とターゲット先端との距離は 35 mm,
基板バイアスは 0 〜 ‑200 V とした.
上記のイオン源に高純度(99.9995%)Ar
ガスを導入し(9 Pa),コイルに RF 電力(260
W)を印加してコイルの内部に Ar プラズマ
を発生させる.金属ターゲットに DC バイア
ス(Cu:‑300 V, Ta:‑500 V)を印加すること
で,ターゲットが Ar+でスパッタされ,金
属イオンと中性粒子が発生し,さらにプラ
ズマ中の準安定 Ar*とのペニング効果[18]
によりイオン化が促進され,多数の金属イ
オンが発生する.金属イオンはコイル上部
の 30 mm φの穴を空けた銅メッシュとの電
位差で引き出され,さらに基板に負のバイ
アス電圧を印加することで加速され,基板に到達する.
成膜したタンタル膜の X 線回折(RIGAKU‑RINT 2000)および Van der Pauw 法によるタンタルの比抵
抗測定を行った.タンタル拡散防止膜の耐熱安定性を調べるため,Cu(100nm)/Ta(50nm)/Si 構造の多
層膜を作製し,常圧の水素雰囲気で 700°C まで 60 分間熱処理を行った.銅薄膜の場合,以前の実験結
果に基づき[16],基板バイアス‑50 V で成膜した.薄膜表面・断面の構造は FE‑SEM(HITACHI‑S‑4100L)
により観察した.
また,650°C で銅の凝集現象が観察されたので,これを抑制する目的で,マグネトロンスパッタ
法により SiO2膜を Cu/Ta/Si 多層膜の上に蒸着し,タンタル膜の耐熱安定性を調べた.マグネトロン
スパッタによる SiO2成膜では 99.99%の純度の SiO2ターゲットを利用し,RF 電圧 500 W を印加し,Ar
雰囲気で SiO2膜を蒸着した.
3. 実験結果および考察
3.1 Ta 膜の比抵抗に及ぼす基板バイアスの影響
作製したタンタル膜の比抵抗およびタンタル膜中のα相とβ相の比の負の基板バイアス依存性を
Fig. 2 に示す.タンタル薄膜ではα相(b.c.c.),β相(tetragonal)そして f.c.c. 相が報告されて
いるが,一般的に蒸着されたタンタル膜では主にβ相になると報告されている[5].また,β相は 110
〜300μΩcm の高比抵抗値を示すが,α相は 24〜50μΩcm の比抵抗でタンタルバルクの値(13μΩcm)
に近いことが知られている[5].Fig. 2 に示したように,基板バイアスを印加しない場合,タンタル
膜は高い比抵抗値(約 250μΩcm)を示したが,負の基板バイアスを増すことで比抵抗値は減少した.
‑125 V の基板バイアスで一番低い比抵抗値(約 40μΩcm)が得られ,以後‑200 V まで徐々に増加する
傾向が見られた.この基板バイアスによる比抵抗値の変化に対しては次の原因が考えられる.まず,
基板バイアスにより,蒸着された薄膜の密度増加と欠陥の減少が挙げられる.銅薄膜の密度と比抵抗
の関係を調べた最近の文献[19]では,銅薄膜の蒸着時,負の基板バイアスを印加することで,薄膜密
度が増加し,‑100 V の基板バイアスで銅バルクに近い比抵抗値が報告されている.本実験でも初期の
比較的低基板バイアス(約‑75 V まで)ではタンタル薄膜の密度増加により比抵抗が減少したと考え
られる.また,基板バイアスによるタンタル膜の相変化が比抵抗に影響を与えたと考えられる.X‑線
林
載元, 裵
俊佑, 三村
耕司, 一色
実
第 59 巻 第 1,2 号
Ratio of α phase/(α+β)
Resistivity (μ Ω㎝)
回折により,αとβピークの強度比から求めた
300
タンタル膜中のα相の比率変化(Fig. 2 の右
スケール)から,‑125 V で蒸着したタンタル
1.0
250
膜はほぼα相である.したがって,相変化が
200
始まる‑75 V から‑200 V までの比抵抗は,主
に相変化によるタンタル自体の比抵抗変化に
150
0.5
起因すると考えられる.
100
そして,もう一つの原因としては基板バ
50
イアスによるタンタル膜中の不純物減少が挙
0.0
げられる.負の基板バイアスを印加すると加
0
0
-50
-100
-150
-200
速されたタンタルイオンは高運動エネルギー
Substrate bias voltage (V)
をもつ状態で基板に衝突し,その時イオン衝
撃(ion bombardment)によるクリーニング効
果が起こる.もし基板表面に弱い結合状態で
Fig. 2 Changes of resistivity and (α/α+β) ratio as a
吸着された不純物があれば,それらの吸着エ
function of the substrate bias voltage.
ネルギーは大略 0.1〜1.0 eV [20],高運動エ
ネルギーを持つタンタルイオンの衝突により表面から除去される可能性が高い.以上,タンタル膜の
比抵抗変化に対する基板バイアスの効果はそれらが相乗的に複合した結果と考えられる.
3.2 Ta 膜の組織に及ぼす基板バイアスの影響
蒸着されたタンタル薄膜の表面と微細構造に及ぼす基板バイアスの影響を観察するため,0, ‑50,
‑125, ‑200 V で作製したタンタル膜を SEM 観察し,Fig. 3 に示す.基板バイアスを印加しない場合
(Fig. 3(a),(b))
,成膜は主にタンタル中性粒子の堆
(a)
(b)
積により進むと考えられ,タンタル原子が基板上で十
分に拡散しないため,柱状結晶成長による柱状構造
(columnar structure)が観察された.さらに表面組
織 では結晶 粒が小 さく,粒 界の隙間 が広く ,亀裂
(c)
(d)
(crack)も観察された.それに比べ,‑50 V の基板バ
イアスを印加した場合(Fig. 3(c),(d))
,結晶粒が確
認できないほど緻密な微細構造を持つことが分かった.
負の基板バイアス印加により,高運動エネルギーを持
つタンタルイオン衝撃により,さらにタンタルイオン
(e)
(f)
の高運動エネルギーが表面での拡散エネルギーに変化
し,タンタル原子の十分な移動によって柱状構造を持
たない緻密な膜ができることが確認された.このイオ
ン衝撃による効果に関して,Hirsch 等は Ge 膜の蒸着
(g)
(h)
時に生じた亀裂によるひび割れ(flaking)などの現象
が,アルゴンイオン照射により無くなったと報告して
いる[21].本実験において基板バイアスを印加した場
300 nm
合,タンタル膜の柱状構造が観察されなかった主因も
イオン衝撃の効果と判断される.
Fig. 3 SEM micrographs of the Ta films
さらに‑125 V の基板バイアスを印加すると(Fig.
at various substrate bias voltages: (a) and
3(e),(f))
,タンタル膜表面の平坦性が若干悪くなり,
(b) 0V, (c) and (d) -50 V, (e) and (f) -125
膜厚も薄くなった.これは蒸着時,過度なイオン衝撃
V, (g) and (h) -200 V.
による逆スパッタ現象によるものと考えられる.さら
平成 15 年 12 月
非質量分離型イオンビームデポジションによる Ta 拡散バリア膜の作製と評価
に,基板バイアスを−200V に増加した場合,逆スパッタ現象はより大きくなり, Fig. 3(g),(h)に示
すように表面が非常に荒れた状態であることが分かる.これまでの結果から,基板バイアス−125V で
作製した Ta 膜はα相でバルクに近い低比抵抗を有すが,表面が若干荒れており,一方,−50V で作製
した Ta 膜は主にβ相で 130μΩcm 程度の高比抵抗であるが,表面は非常に平坦であることが分かった.
これらの結果を基に,次節の Cu/Ta/Si の熱安定性を調べる際には,負の基板バイアス 0V,−50V,−
125V で作製した Ta 膜をバリア層として用いた時の得失を検討した.
Resistivity (μ Ω㎝)
3.3 Cu/Ta/Si 多層膜の耐熱安定性
Cu(100nm)/Ta(50nm)/Si 構造の多層膜を作製し,この多層膜の熱安定性に及ぼす Ta 拡散防止膜作
製時の基板バイアスの影響を調べた.Fig. 4 は室温から 700℃で水素雰囲気中にて熱処理した後の多
層膜の比抵抗を示す.基板バイアスを印加しない
場合,Cu/Ta/Si 多層膜の比抵抗は 400°C で急激な
200
(a)
増加が見られた.この原因に関しては,後で詳細
(b)
に検討するが,銅原子がタンタル拡散防止膜内の
(c)
150
結晶粒界に沿って拡散し,シリコン側でシリコン
と反応し,化合物を形成したことに起因すると考
100
えられる.一方、基板バイアス−50V−125V を印加
し,Ta 拡散防止膜を作製した場合,両方とも 600℃
50
まで比抵抗の増加は見られず,650〜700℃で若干
の比抵抗の増加が認められた.なお,前述した通
0
り,基板バイアスの相違により Ta 膜はα相(−
0
100 200 300 400 500 600 700
50V),β相(−125V)と異なるが,熱処理温度に対す
Temperature (℃)
る多層膜の比抵抗変化はほぼ同様な傾向を示し,
Ta 膜の相構造は耐熱安定性に影響しないと判断
Fig. 4 Resistivity changes as a function of
される.したがって,基板バイアスによるイオン
annealing temperature in H2 for 60 min.: (a) the
衝撃の増大により Ta 膜が緻密化され,その結果銅
Cu/Ta(0V)/Si, (b) the Cu/Ta(-50V)/Si and (c) the
原子の拡散が抑えられ,多層膜の耐熱安定性が向
Cu/Ta(-125V)/Si samples.
上したと考えられる.そこで,以上の結果を詳細
に検証するため,熱処理前後の Cu/Ta/Si 多層膜内の変化を XRD および SEM 観察で調べた.先ず,
Cu/Ta(V)/Si 多層膜の熱処理前後の XRD 結果および 400℃で熱処理した多層膜の表面と断面組織を Fig.
5 に示す.XRD 結果から,300℃までは大きな変化も無く安定しているが,400℃では Cu ピークが消え
Cu3Si のピークが確認された.また Fig. 5(b)から円形状の膨らみが表面に出現し,Fig. 5(c)の断面
観察から Ta 膜上の Cu 膜がほぼ完全に消失したことが分かる.これは Ta 膜が柱状構造(Fig. 3(b))を
有することから,400℃程度の低温熱処理でも,Cu 原子が Ta 膜の結晶粒界に沿って Si 側に拡散し,
Ta/Si 界面で Si と反応し,Cu3Si を生成したと考えられる.生成した Cu3Si は約 150%膨張することが
報告されており[22],Fig. 5(c)からも明らかなように,表面の円形状の膨らみは Cu3Si 生成に起因し
たものである.
一方、基板バイアスを印加して作製した Ta 膜の場合は,全く異なる挙動を示した.Fig. 6,7 に
Cu/Ta(‑50V)/Si と Cu/Ta(‐125V)/Si 多層膜の熱処理前後の XRD 結果および 700℃熱処理後の多層膜の
表面と断面組織を示した.両方とも 600℃までは XRD に大きな変化は無く安定し,比抵抗の結果(Fig.
4)に対応し,組織観察からも耐熱安定性が向上していることが確認された.しかし,650℃では Cu ピ
ークが急速に小さくなり,CuTa10O26 ピークが出現し,700℃では CuTa10O26 ピークが増大した.700℃熱
処理後の観察結果(Fig. 6(b),(c)と Fig. 7(b),(c))から,
円形島状の物質が Ta 膜上に認められたが,
X 線分析マップから Cu の凝集物であることが確認された.銅が薄膜化された場合,融点より低い温度
でも銅原子の移動(拡散)により凝集することが知られている.この銅薄膜の凝集現象は最近多く報告
林
載元, 裵
俊佑, 三村
耕司, 一色
第 59 巻 第 1,2 号
実
(b)
(111) Cu
(200) Cu
(200) β-Ta
Cu3Si
Intensity (a.u.)
(a)
400℃
500 nm
300℃
200℃
as deposited
20
30
40
50
60
2θ
Fig. 5 (a) XRD patterns of the Cu/Ta(0V)/Si sample as
a function of annealing temperature and (b) surface
and (c) cross-sectional SEM micrographs of the
sample after the annealing at 400°C.
(c)
Ta
Cu3Si
300 nm
され,実際のデバイス製造工程で高温熱処理を必要とするメモリー分野などで問題化しており,改善
が求められている[23‑25].上記の実験結果で,Ta 成膜時の負の基板バイアス印加により耐熱安定性
が向上したのは,Fig. 6(c)と 7(c)の断面写真からわかるように,バリア相の Ta 膜内に柱状構造が観
察されず緻密な微細構造とイオン衝撃による欠陥減少による効果と考えられる.
650℃における熱処理後に XRD で確認された CuTa10O26 は,CuO と 5Ta2O5 で構成された複合酸化物と
考えられるが,以下その形成過程を検討した.先ず酸素の供給源としては,銅酸化層(CuO,Cu2O),SiO2,
熱処理時の水素ガス中の酸素,水分などを考える必要があるが,さらに熱処理後試料を大気暴露した
際の酸化も考えられる.この中で,銅酸化層は水素熱処理時に銅に還元されるから排除でき,SiO2 中
の酸素も 900℃まで TaSiO2 膜が安定に存在する[26]ことが知られており,考慮外である.したがって,
熱処理時の残留酸素・水分と大気暴露時の酸化が主因と推測される.そこで,650℃における平衡酸素
ポテンシャルを求めてみると[27],
Cu/Cu2O の場合 PO2 は約 4.1×10‑7Pa、
Ta/Ta2O5 では PO2 は約 1×10‑32Pa
であり,極低酸素ポテンシャルで容易に Ta が酸化されることが判る.なお,水素熱処理時 650℃にお
ける PO2 は 2×10‑30Pa 程度と予測され,Ta 膜が熱処理時に露出した場合酸化される可能性が高い.し
たがって,今回の結果から,650℃の熱処理時に銅の凝集が観察され,Ta 膜が露出したことから,水
素熱処理中でも酸化が生じ,さらに大気暴露時に酸化が加速されたと考えられる.また,凝集した銅
も大気暴露時に酸化層が形成され,銅酸化層(CuO),と Ta 酸化層(Ta2O5)により CuTa10O26 が形成された
ものと考えられる.
負の基板バイアスを印加し作製した Ta 膜の場合,650℃熱処理後の多層膜の比抵抗上昇は,銅薄
膜の凝集が主因であり,Ta 膜自体の問題ではないことが明らかにされた.そこで Cu/Ta/Si 多層膜上
に SiO2 膜を蒸着し,銅薄膜の凝集を抑えた状態で,Ta 拡散防止膜の耐熱安定性を再検討した.Fig. 8
に,SiO2/Cu/Ta(‑125V)/Si 多層膜を 650℃と 700℃で熱処理後,SEM 観察した表面および断面の微細構
造を示す.Fig. 8(a),(b)から,熱処理温度 650℃で SiO2 膜の存在により銅の凝集は見られず,Ta 拡
散防止膜も効果的に銅の拡散を抑制し,基板バイアスを印加し作製した Ta 膜が 650℃まで拡散防止膜
として有効に働くことが確認された.この結果は,表 1 に示した報告値との比較から,Ta 拡散防止膜
として最高温度域での耐熱安定性が得られたことになる.なお,Fig. 8(c),(d)から,熱処理温度 700℃
では Cu3Si が形成され,基板バイアス印加なしの場合と同じく,その膨張により表面に円形状の膨ら
みが生じ,700℃では熱安定性の維持が困難なことが判った.
平成 15 年 12 月
非質量分離型イオンビームデポジションによる Ta 拡散バリア膜の作製と評価
(111) Cu
(200) Cu
(200) β-Ta
(110) α-Ta
CuTa10O26
Intensity (a.u)
(a)
(b)
700℃
1 µm
650℃
600℃
500℃
as deposited
20
30
40
50
60
2θ
(c)
Cu
Fig. 6 (a) XRD patterns of the Cu/Ta(-50V)/Si
sample as a function of annealing temperature and
(b) surface and (c) cross-sectional SEM micrographs
of the sample after the annealing at 700°C.
(b)
(111) Cu
(200) Cu
(200) β-Ta
(110) α-Ta
CuTa10O26
Intensity (a.u)
(a)
300 nm
Ta
700℃
1 µm
650℃
600℃
500℃
20
30
40
50
60
as deposited
2θ
(c)
Cu
Fig. 7 (a) XRD patterns of the Cu/Ta(-125V)/Si
sample as a function of annealing temperature and (b)
surface and (c) cross-sectional SEM micrographs of
the sample after the annealing at 700°C.
Ta
300 nm
林
載元, 裵
俊佑, 三村
(a)
耕司, 一色
第 59 巻 第 1,2 号
実
(b)
SiO2
Cu
Ta
3 µm
(c)
200 nm
(d)
SiO2
Cu3Si
5 µm
Si
3 µm
Fig. 8 SEM micrographs of the SiO2/Cu/Ta(-125V)/Si sample after annealing: (a) and (b) at 650°C, (c)
and (d) at 700°C.
以上の結果から,Ta 膜蒸着時に負の基板バイアスを印加することで,より緻密な微細構造を有す
る Ta 拡散防止膜が形成され,多層膜の耐熱安定性が向上することが明らかにされた.
結言
非質量分離型 IBD 装置を用い,Cu/Ta/Si 多層膜のおける Ta 拡散バリア膜を作製し,特に Ta 膜作
製時の負の基板バイアス印加による効果を検討し,多層膜の耐熱安定性に及ぼす Ta 拡散バリア膜の影
響を評価した.得られた結果の要約を以下に記す.
1)基板バイアスを印加しない場合,Ta 膜は約 250μΩcm の高比抵抗値を示すが,負の基板バイス増
加に伴い,Ta 膜の緻密性が向上し比抵抗値の低下が確認された.基板バイアス−125V で最少の比抵抗
値約 40μΩcm が得られたが,以後−200V まで比抵抗値は若干増加する傾向を示した.
2)Cu/Ta/Si 多層膜の耐熱安定性に関し,Ta 膜作製時で基板バイアスなしの場合,400℃で Ta 膜の粒
界に沿って Cu 原子が拡散し,Ta/Si 界面で Cu3Si が形成され,多層膜の比抵抗が増大した.一方,負
の基板バイアス印加し作製した Ta 膜の場合,650℃まで Cu の拡散が効果的に抑制され,多層膜の耐熱
安定性の向上が確認された.
3)負の基板バイアス印加により作製された Ta 膜は,柱状構造が観察されず緻密な微細組織を有し,
これはイオン衝撃による Ta 膜内の欠陥が減少するためと考えられる.
4
文献
(1) 吉川公麿:応用物理, 68(1999), 1215.
(2) P. Murarka; R. J. Gutmann; A. E. Kaloeros; W. A. Lanford: Thin Solid Films, 236(1993), 257.
(3) M. Proust; F. Judong; J. M. Gilet; L. Liauzu; R. Madar: Microelectron. Eng., 55(2001), 269.
(4) 守山実希,村上正紀:応用物理, 68(1999), 1247.
平成 15 年 12 月
(5)
(6)
(7)
(8)
非質量分離型イオンビームデポジションによる Ta 拡散バリア膜の作製と評価
P. N. Baker: Thin Silid Films, 14(1972), 3.
C. A. Chang: J. Vac. Sci. Technol. A, 8(1990), 3796.
E. Kolawa; J. S. Chen; J. S. Reid; P. J. Pokela; M. A. Nicoler: J. Appl. Phys., 70(1991), 1369.
K. Hollowas; P. M. Fryer; C. Cabral Jr.; J. M. E. Harper; P. J. Bailey; K. H. Kelleher: J. Appl. Phys.,
71(1992), 5433.
(9) A. Ohta; A. Noya; M. Takeyama; M. Taguchi; T. Sase; K. Sasaki: Thin Solid Films, 278(1996), 6.
(10) J. S. Kwak; H. K. Baik; J. H. Kim; S. M. Lee: Appl. Phys. Lett.. 71(1997), 2451.
(11) A. Z. Moshfegh; O. Akhavan: Thin Solid Films, 370(2000), 10.
(12) J. Amano; P. Bryce; R.P. W. Lawson: J. Vac. Sci. Technol., 12(1976), 591.
(13) T. Tsukizoe; T. Nakai; N. Ohmae: J. Appl. Phys., 48(1977), 4770.
(14) M. Yamashita: Jpn. J. Appl. Phys., 26(1987), 721.
(15) K. Miyake; K. Ohashi: Nucl. Instr. Meth. B, 121(1997), 102.
(16) J. -W. Lim; Y. Ishikawa; K. Miyake; M. Yamashita; M. Isshiki: Mater. Trans., 43(2002), 1403.
(17) K. Miyake; Y. Ishikawa; M. Yamashita; M. Isshiki: Proceedings of 2000 International Conferences on Ion
Implantation Technology, Alpbach, Austria, (2000), 550.
(18) J. W. Coburn; Eric Kay: Appl. Phys. Lett., 18(1971), 435.
(19) H. M. Choi; S. K. Choi; O. Anderson; K. Bange: Thin Solid Films, 358(2000), 202.
(20) T. Takagi: Thin Solid Films, 92(1982), 1.
(21) E. H. Hirsch, I. K. Varga: Thin Solid Films, 52(1978), 445.
(22) M. Seibt; M. Griess; A. A. Istratov; H. Hedemann; A. Sattler; W. Schröter: Phys. Stat. Sol. A, 166(1998),
171.
(23) T. Hara; K. Sakata: Electrochem. Solid-State Lett., 4(2001), G77.
(24) J. J. Rha; J. K. Park: J. Appl. Phys., 82(1997), 1608.
(25) T. Ichinokawa; H. Izumi; C. Haginoya; H. Itoh: Phys. Rev. B, 47(1993), 9654.
(26) S. Q. Wang; J. W. Mayer: J. Appl. Phys., 67(1990), 2932.
(27) O. Kubaschewski; C. B. Alcock: Metallurgical Thermochemistry, 5th ed., (1979), 378, Pergamon Press.
胴長の異なる転動ミル内における媒体運動と粉砕速度との相関
三尾
浩*,加納
純也*,齋藤
文良*
Correlation of Grinding Rate with Specific Ball Impact Energy Simulated in Tumbling Mills
with Different Pot Lengths
By Hiroshi MIO, Junya KANO and Fumio SAITO
Dry grinding of a gibbsite was conducted using tumbling ball mills with different pot lengths to investigate
its effect on the grinding rate determined by the experiment. The ball motion in the mill was simulated by
DEM, and the specific impact energy of balls was calculated. The grinding rate increases with an increase in
the rotational speed of the mill, and falls rapidly around the critical speed. As the mill length becomes short,
the grinding rate becomes large due to the interaction between balls and the side-wall of the pot. All the same,
the grinding rate is expressed by a linear function with the specific impact energy regardless of the pot length.
Therefore, the grinding rate in tumbling ball milling is predicted by the specific impact energy of balls under
any conditions.
(Received October 10th, 2003)
Keywords: grinding rate, Discrete Element Method, specific impact energy, tumbling ball mill, size reduction
緒言
粉砕操作は固体の比表面積を大きくするだけでなく,その反応性を向上させたり,含有有価成分の
単体分離や,固体の搬送などの操作性を容易にするなど,多くの目的を持っている.したがって,こ
の操作は化学工業をはじめ,医薬,食品工業など,多くの産業で取り入れられている.近年は,資源
の枯渇問題や環境保全への取り組みが重視される中,廃棄物処理やリサイクルが盛んに行われている
が,ここでも粉砕処理が多く取り入れられ,かつては鉱石処理で多用されていた操作が,この分野で
再び日の目を見始めている.
代表的な粉砕機として,媒体(ボール)を利用する各種ミルがあるが,その中でも,容器(ポット)
回転型の転動ミルは,ポットに自転運動を与えるだけの単純な動作機構で,大量処理が可能であるた
め,多用されている.しかしながら,粉砕機は一般にエネルギー効率は低く,その僅かな効率(粉砕
速度など)向上でも,長時間運転では積算で効いてくるため経済効果は無視できず,したがって,そ
のための運転条件やミル構造の最適化が強く望まれ,この点は転動ミルでも同様である.これまで,
転動ミルにおける粉砕速度(効率)向上に関する研究は,実験的には多く行われている 1-3 が,大きさ
が変わればその都度実験によって最適条件を見出す必要があり,普遍的な最適条件設定法は無かった.
その最大の理由は,転動ミル内における媒体の運動を正確に把握する手段・ 手法が無く,中で起こっ
ている現象がいったいボール群のどのような運動因子によって支配されているかを見極めることがで
きていなかったことによる.もちろん,粉砕速度論などは盛んに検討され,マクロ的な現象把握はあ
ったが,ここのボール運動の把握という粉砕に直接関連するミクロな議論ではなかった.ところが,
近年,DEM (離散要素法,Discrete Element Method)4 を利用して,粒子群軸運動の解析ができるように
1.
* 東北大学多元物質科学研究所
平成 15 年 12 月
胴長の異なる転動ミル内における媒体運動と粉砕速度との相関
なり,これをミル内ボール群運動の解析に利用する試みが多くなってきた.その結果,これまでブラ
ックボックスとみなされてきたミル内の媒体運動が時間的,空間的に解明され,媒体運動が克明に解
析できるようになった 5-7.しかしながら,このようにしてシミュレーションされたミル内ボール運動
と実験結果を結びつけ,相互の相関性を諮る研究は依然として少ないのが現状である.
加納らは,実験とシミュレーションの接点の重要性に着目し,両者の相関性を明確にしつつ,シミ
ュレーションからの情報を基にして実験結果を予測することに成功した.すなわち,砕料がミル内に
存在している条件を模擬したボール運動のシミュレーション法 8 を開発し,それらの運動を明確にし
つつ,一方では実験によって砕料の粉砕速度を,ミル径 9,ボール充填率 10,ボール径 11 の異なる条
件下において実測し,これらとシミュレーション結果との相関を試み,粉砕速度に最も影響するパラ
メータは媒体衝突エネルギーであることを見出している.
本報告では,これまで未検討であった転動ミルの胴長を種々変化させた場合のボール運動の影響と
粉砕速度との関連性を検討することにした.
実験
ミル胴長lMは(lM =71, 142, 213mm,直径121mm一定)3段階に変化させた.また用いたボールは,
径が10.2, 15.9, 19.1mmの3段階に変化させたものであり,それらを見かけ充填率で40%となるように
充填した.砕料(被粉砕試料)には,初期平均粒子径33.3μmのギブサイト(Al(OH)3, CHP340S, 住友
化学工業(株))を用いた.この試料をポット容積に対して20%となるように充填し,ミル回転速度は式
(1)で定義される臨界回転速度(NC)の0.6〜1.2倍に設定して,所定時間粉砕後,産物試料を少量サンプ
リングして粒子径をレーザー散乱式粒度分布測定器(Laser Micronsizer LMS‑30, セイシン企業)に
より測定後,再び粉砕を繰り返した.詳細な実験条件を表1に示す.
2.
NC =
60
π
g
2d M
(1)
ここで,gは重力加速度,dMはミル径である.
Table 1 Experimental condition
Pot diameter
Pot length
Ball diameter
Ball-filling ratio
Number of ball
Sample loading
Critical rotational speed
d M [mm]
121
l M [mm] 72
142 215
72
142 215
72
142 215
d B [mm]
10.2
15.9
19.1
40
J
[%]
nB
[-] 317 624 949
84
167 252
49
97
146
W
[kg] 0.152 0.300 0.456 0.152 0.300 0.456 0.152 0.300 0.456
N C [rpm]
121.5
シミュレーション
DEMは個々の粒子(ここでは媒体を意味する)の運動方程式を導入することにより離散的に粒子運動
を解析するシミュレーション法であり,2つの粒子間の衝突は図1に示されるスプリング(弾性)と
ダッシュポット(粘性)で構成されるVoigtモデルで表される.ボール間接触力Fn(圧縮), Ft(せん
断)は式(2),(3)で与えられる.
3.
三尾
Fn = K n ∆ u n + η n
第 59 巻 第 1,2 号
浩, 加納純也, 齋藤文良
∆u n
∆t
(2)
∆ (u t + r ϕ )
Ft = min µ F n , K t ∆ (u t + r ϕ ) + η t
∆t
(3)
ここで,u, φはそれぞれ着目2粒子間の相対変位,相対回転変位であり,K, η, μは,ばね定数,
粘性係数,摩擦係数を表す.シミュレーションで用いたミルのサイズや操作条件は,実験における粉
砕条件と同じとし,ヤング率,ポアソン比はそれぞれ210GPa,0.30,摩擦係数は被粉砕物の安息角か
ら0.68と決定した8.なお,ミル内のボールの運動は1μsec毎に計算を進めた.
2ボール間,あるいはボール−ミル壁間における衝突速度はシミュレーションから求められ, その時
の衝突エネルギーは相対速度とボールの特性などから式(4)で算出した.ここにm, n は,それぞれ媒
体質量と1秒間の衝突回数を表し,Wは充填した試料質量である.
EW =
M =
n
∑
j =1
1
Mv
2W
2
r,j
(4)
2 m1 m 2
m1 + m 2
(5)
m1
uunn
m1
ϕ
Kt
uutt
ηn
slider
Kn
ηt
m2
µ
m2
(a) Compressive force
(b) Shear force
Figure 1 Voigt model.
結果および考察
図2には直径が10.2mmのボールを使用して3種類の胴長のミルで,回転速度=121.5rpm (N/NC = 1.0)
で粉砕した時の砕料の平均粒子径の経時変化を示す.粒子径はすべて初期粒子径で規格化しており,
図中の曲線は式(6)で近似したものである.
4.
Dt
D
D
= 1 − l exp (− K P ⋅ t ) + l
D0
D0
D0
(6)
胴長の異なる転動ミル内における媒体運動と粉砕速度との相関
1.0
Normalized 50% particle size, Dt/D0 [-]
ここで,Dは平均粒子径,添字t, l , 0はそ
れぞれ,粉砕時間t,粉砕限界,未粉砕を表
す.式(6)におけるKPを粉砕速度と定義する.
粒子径は粉砕時間と共に指数関数的に減少
する一方 , ミル胴長lMの影響は非常に小さ
いことがわかる.図3には直径が10.2mmの
ボールを用いたときのKPとN/NCの関係を示
す . KPはミルの回転速度の増加に伴って大
となり , N/NC = 0.8のところで極大値をと
り,それ以上の回転速度では急激に減少す
る.この傾向はミル胴長の異なるポットで
もほぼ同じである.但し,胴長の狭いポッ
トでは,粉砕速度は極大値を示した後の減
少量が大きい.これは,ミル胴長が狭いた
め,ボールとポット側壁との相互作用が大
きくなったためであって,共廻り現象が現
れためと考えられる.このことは,シミュ
レーションにおけるボール運動からも確認
された.図4には,シミュレーションから
得られたボール運動のスナップショットを
示す.ボールの上層表面の傾きは大きく,
側壁とボールとの接触の影響が大きいこと
がわかる.一方, lM = 213 mmにおいては,
KPはピーク値を示した後 ,緩やかに下降し
ている.これは, lM = 71 mmの場合とは逆
に,ミル胴長が大きいため,ミル壁に影響
されないボールの数が多く,ミル壁とボー
ルとの相互作用を持つボールの割合が少な
いことによる(図4)
.
図5には ,ボールの衝突エネルギーEWと
ミル回転速度比(N/NC)との関係を示す . EW
は(N/NC)の増加に伴い大となり , NCの80 〜
90%で極大値を示した後,急激に減少する
傾向にある.さらに, lM = 71 mmのミルで
は,側壁の影響が大きいため共廻りが出現
しやすく , したがってEW値が小さくなって
いることがわかる.この傾向は実験で得ら
れたものとよく対応し,さらに,直径が15.9,
19.1mmのボールを用いた場合でも同様であ
った.これより, EWはミルの粉砕能を表す
良いパラメータと言える.
実験によって得られた砕料のKPと , EWの
関係を図6に示す.プロットされた点は,
Pot length
71mm
142mm
213mm
0.8
0.6
0.4
0.2
0.0
0
2000 4000 6000 8000 10000 12000
Grinding time, t [sec]
Figure 2 Relation between the normalized 50%
particle size and the grinding time.
1.0x10-3
8.0x10-4
Grinding rate, KP [s-1]
平成 15 年 12 月
6.0x10-4
4.0x10-4
2.0x10
-4
Pot length
71
142
213 mm
0.0
0.5 0.6 0.7 0.8 0.9 1.0 1.1 1.2 1.3
Relative rotational speed, N/NC [-]
Figure 3 Relation between the grinding rate and the
relative rotational speed.
三尾
第 59 巻 第 1,2 号
浩, 加納純也, 齋藤文良
(a) lM = 71 mm
(b) lM = 142 mm
(c) lM = 213 mm
Figure 4 Snapshots of the side view under three conditions of different pot length.
50
Specific impact energy, EW [J/(s.kg)]
分散しているが,ほぼ1本の直線上に集中し
ており , したがってEWとKPの間には比例関
係があることが分かる.また,この関係は
ミル胴長,ボール径に関わらず,同一の直
線近傍に分散していることがわかった.し
かし,高回転速度下では,データは直線か
ら少しはずれている.これはボールの共廻
り現象が現れた時に,実験では媒体が大量
に存在する粉体中に埋まり,粉砕が全く進
行しなくなるという現象が起こるが,シミ
ュレーションでは,このことを正確に表現
できないためである.なお,前述したよう
に,筆者らは,ギブサイトの転動ミル粉砕
において,ミル径9,ボール充填率10,ボー
ル径11を変化させたときのボール運動のシ
ミュレーションを行い , EWとKPの関係を検
討してきたが,ここで検討した胴長を加え
た4つのパラメータを変化させたときのEW
40
30
20
Pot length
71
142
213mm
10
0
0.6
0.7
0.8
0.9
1.0
1.1
1.2
Relative rotational speed, N/NC [-]
Figure 5 Relation between the specific impact energy
and the relative rotational speed.
胴長の異なる転動ミル内における媒体運動と粉砕速度との相関
(7)
結言
本研究では,胴長(ミル長さ方向の距
離)の異なる転動ミルによるギブサイト
粉末の乾式粉砕から,その粉砕速度を実
測し , 一方ではミル内ボール運動をDEM
に基づくコンピュータシミュレーション
法からボールの衝突エネルギーを求め,
両者の関連性を検討した.その結果,次
の知見を得た.
(i) 粉砕速度はミル回転速度の増加に
伴って大きくなり , 臨界回転速度
の約80%のところで最大となった
後減少する . この臨界回転数以上
の条件では , ミル胴長の狭い場合
は , 側壁からボール運動が受ける
影響が大きく , ボールは共廻りし
やすくなり , 砕料の粉砕速度は急
激に減少する . 逆に , この回転速
度域において , ミル胴長が広い場
合は , 側壁のボール運動への影響
が小さく , したがって , 粉砕速度
は緩やかに減少する.
(ii) コンピュータシミュレーション法
より算出されるボール衝突エネル
ギーは , 実測の砕料の粉砕速度と
良好に相関し , 両者の間には一定
の関係が成り立つ . さらに , ここ
で得られた関係は , これまでに検
討してきたミル径,ボール充填率,
ボール径の影響を明確にした実験
と同じであり , すべて一本の直線
で表されることを明確にした . こ
のことは , 転動ミルにおけるギブ
5.
8.0x10-4
Grinding rate, KP [s-1]
K P = kEW
1.0x10-3
6.0x10-4
4.0x10-4
2.0x10-4
0.0
0
10
20
30
40
50
Specific impact energy, EW [J/(s.kg)]
Figure 6 Relation between the grinding rate and the
specific impact energy.
lM=71mm ; [dB=10.2], [dB=15.9], [dB=19.1]
lM=142mm; [dB=10.2], [dB=15.9],[dB=19.1]
lM=213mm; [dB=10.2], [dB=15.9],"[dB=19.1]
2.0x10
-3
1.5x10
-3
1.0x10
-3
5.0x10
-4
-1
との間には良好な相関関係が成り立ち,
その関係は式(7)で与えられるといえる.
ここに,kは被粉砕試料の材料定数や初期
粒子径に依存する定数12であり . この式
を用いると,これまでの検討結果より同
一砕料(ギブサイト)の場合は,シミュ
レーションより得られるボール運動エネ
ルギーからその砕料の粉砕速度が推算で
きるものと考えられる.
Grinding rate, KP [s ]
平成 15 年 12 月
0.0
0
20
40
60
80
Specific impact energy, EW [J/(s.kg)]
Figure 7 Correlation between the grinding rate and the
specific impact energy under all parameters.
三尾
浩, 加納純也, 齋藤文良
第 59 巻 第 1,2 号
サイトの粉砕に限定して , シミュレーション法で求められるボールの衝突エネルギーから , こ
の砕料の粉砕速度が推算できる可能性が大であり , ミルのスケールアップ基準が確立できるも
のと思われる.
References
(1) R.E. McIvor, Mining Eng., 35 (1983), 617.
(2) H.J. Steiner, Int. J. Miner. Process. 44‑45 (1996), 373.
(3) 伊藤光弘,長野健一,樋野公彦,化学工学論文集, 22 (1996), 503
(4) P.A Cundall and O.D.L. Strack, Geotechnique, 29 (1979), 47.
(5) B.K. Mishra and R.K. Rajamani, KONA, 8 (1992), 92.
(6) P.W. Cleary, Miner. Eng., 11 (1998), 1061.
(7) M.A. van Nierop, G. Glover, A.L. Hinde and M.H. Moys, Int. J. Miner. Process., 40 (2001),
77.
(8) J. Kano, N. Chujo and F. Saito, Adv. Powder Technol., 8 (1997), 39.
(9) J. Kano, H. Mio, F. Saito and M. Tanjo, J. Chem. Eng. Jpn, 32 (1999), 747.
(10) J. Kano, H. Mio and F. Saito, AIChE J., 46 (2000), 1694.
(11) J. Kano, H. Mio, F. Saito and M. Miyazaki, Miner. Eng., 14 (2001), 1213.
(12) J. Kano, H. Mio and F. Saito, J. Chem. Eng. Jpn, 32 (1999), 445.
集束イオンビーム法と電子線ホログラフィーを用いた
Fe-Co-Cu-Nb-Si-B ナノ結晶磁性材料の磁区構造評価
斉藤健太*, 朴英吉**, 村上恭和**, 進藤大輔**, 吉沢克仁***
Analysis of Magnetic Domain Structure in Fe-Co-Cu-Nb-Si-B Nanocrystalline Magnetic
Materials by Focused Ion Beam Method and Electron Holography
By
Kenta SAITO, Young-Gil PARK, Yasukazu MURAKAMI, Daisuke SHINDO and
Yoshihito YOSHIZAWA
Magnetic domain structures of Fe-Co-Cu-Nb-Si-B alloys, in which the uniaxial anisotropy is induced by the
magnetic annealing, were investigated by Lorentz microscopy and electron holography. While the specimen
prepared by the ion-milling showed a rather complicated magnetic domain structure due to the drastic change in
specimen thickness, the specimen prepared by the focused ion beam method, which had a uniform thickness,
showed a characteristic domain structure induced by the magnetic anisotropy. The magnetic domain width in
Fe18.8Co60Cu0.6Nb2.6Si9B9 was smaller than that in Fe78.8Cu0.6Nb2.6Si9B9, and this result was reasonably
explained by the magnitude of the magnetic anisotropy. The magnetic flux density (1.6 T) measured by
holography for Fe78.8Cu0.6Nb2.6Si9B9 was consistent with that observed for a bulk specimen (1.5 T).
(Received October 10th, 2003)
Keywords: focused ion beam method, electron holography, the lines of magnetic flux, magnetization distribution
緒言
近年、透過型電子顕微鏡(以下、電顕と略す)に、高輝度でかつ干渉性の高い電子線が得られる電
界放出型電子銃(Field Emission Gun : 以下 FEG)が搭載され、ナノオーダーでの構造解析に加え電子
波の干渉実験が可能になっている。この電子波干渉実験法の一つである電子線ホログラフィーでは、
結晶下面での電子の位相情報を再生することにより、内部ポテンシャルや磁区構造、磁化分布など様々
な情報が得られてきている 1)。現在、この電子線ホログラフィーの各種先端磁性材料の磁区構造評価
への応用に大きな期待が寄せられている。
Fe ベース(Fe-Cu-Nb-Si-B)のナノ結晶磁性材料は高透磁率や低コアロスなど興味深い磁気特性を示
しており、ノイズ対策部品やパワーエレクトロニクス関連部品、情報通信機器部品などに応用されて
いる 2) 。これらに要求される周波数特性は近年さらに高周波数側に移行している。このため、
Fe-Cu-Nb-Si-B 合金に Ni あるいは Co 元素の添加と磁界中結晶化による誘導磁気異方性を用いること
で高周波領域で優れた軟磁性特性を実現するための研究がなされている 3)。磁性材料における磁気特
性は微細構造と磁区構造に深く関わっていることが知られており、現在多くの研究が行われている 4)-6)。
これまでの磁区構造評価には、形状や厚さの制御は困難であるが広い範囲において薄い領域を得る
ことができるイオンミリング法で作製された試料を用いて行われている。しかしながら軟磁性材料の
磁区構造は試料の形状と厚さの影響を非常に受けやすいため 7)8)、これらの因子を制御するための実験
1.
*
東北大学大学院工学研究科
** 東北大学多元物質科学研究所
*** 日立金属株式会社 先端エレクトロニクス研究所
第 59 巻 第 1,2 号
斉藤健太, 朴英吉, 村上恭和, 進藤大輔, 吉沢克仁
Table 1 Uniaxial anisotropy constant Ku and magnetic flux density B of Fe-Co-Cu-Nb-Si-B alloys.
組成(原子%)
試料 A Fe78.8Cu0.6Nb2.6Si9B9
試料 B Fe18.8Co60Cu0.6Nb2.6Si9B9
Ku(Jm-3)
B(T)
96
1.5
1143
1.2
Fig.1 Scanning ion microscope images showing specimen preparation by the focused ion beam (FIB) method: deposition
of carbon on the specimen surface (a), coarse polishing to obtain a thin film (b), (c), final stage of polishing using a finer
ion probe (d). Pairs of white arrows indicate a specimen thickness at each stage. A plan-view image of the film
(bright-field image) is shown in the inset of (d).
法が望まれている。一方、イオンビームを微小領域に絞り、試料表面をスキャンさせながらスパッタ
する集束イオンビーム法(以下、FIB 法)は、最近ナノメータ−オーダーの均一な厚さと形状が揃った電
顕観察用試料が得られる手法として注目を集めている。本研究では、FIB 法により試料を作製し、試
料 内 部 での 磁 化分 布 を 評価 で き る電 子 線ホ ロ グ ラフ ィ ー を用 い 誘導 磁 気 異方 性 が 誘起 さ れた
Fe78.8-xCoxCu0.6Nb2.6Si9B9(x=0、60)の磁区構造評価を行った。
実験方法
本研究に用いた試料は、液体急冷法により作製された Fe78.8Cu0.6Nb2.6Si9B9 (ここでは試料 A と呼ぶ)
合金リボンと Fe18.8Co60Cu0.6Nb2.6Si9B9(試料 B)合金リボンを、それぞれ窒素ガス雰囲気中でリボンの幅
2.
平成 15 年 12 月
集束イオンビーム法と電子線ホログラフィーを用いた
Fe-Co-Cu-Nb-Si-B ナノ結晶磁性材料の磁区構造評価
方向に磁界をかけながらナノ結晶化したものであり、これらの磁気特性は Table 1 に載せてある。これ
らをイオンミリング装置(MODEL 600NDIF )および FIB 装置(JEM-9310FIB)にて加工した。
主な試料作製の手順としては、まずリボンの幅方向が観察用試料の端とほぼ垂直になるように予め
切り出して、それぞれ Mo メッシュ(イオンミリング法)および Mo 半メッシュ(FIB 法)に貼り付けた。
イオンミリング法では、アルゴンイオンを用いて 3kV、1mA の条件で小さい穴があくまで研磨し電顕
試料とした。FIB 法では、試料の端面を保護するためにカーボン蒸着を施した。その様子を Fig.1(a)
に示す。その後、30kV で加速した Ga イオンを用いて粗加工 Fig.1(b)、中加工 1(c)、仕上げ加工 1(d)
と進めて行き、最終的に約 60nm 程度の厚さにした。Fig.1(d)の挿入図に FIB 法で作製した試料の電子
顕微鏡像(電子線は試料の膜面と垂直に入射)を示す。この図から、作製した試料の端部が平行に制御
されていること及び一様なコントラストから膜厚がほぼ均一になっていることがわかる。
磁区構造の観察には、加速電圧 300kV で FEG とバイプリズム(Biprism)を搭載した JEM-3000F 電顕
を用いた。試料位置での残留磁場は 0.2mT 程度である。ホログラムの撮影は、倍率 3400 倍およびバ
イプリズム電圧 35kV の条件で行なった。記録媒体には、従来の電顕フィルムを用いた。電顕フィル
ムに記録したホログラムは、フィルムスキャナー(MINOLTA DiMAGE Scan Multi PRO)を用いてディジ
タルデータに変換し、画像解析(高速フーリエ変換)を用いて、磁束線の情報が得られる位相再生像を
得た。
結果および考察
磁性材料の評価において、イオンミリング法により作製された試料は広い電顕観察領域を得られる
一方で形状や厚さの制御が難しく、形状の影響を受けやすい軟磁性材料においては違う組成の材料を
直接比較することは難しい。これに対し、FIB 法により作製された試料は形状や厚さを制御できるた
め、これらによる磁区構造への影響を小さくすることが出来る。リボンの幅方向に高い誘導磁気異方
性を持つ試料 B に対して、EELS(electron energy – loss spectroscopy)による相対試料厚さの評価と電子顕
微鏡観察を行なった。その結果を Fig.2 (a)、(b) (イオンミリング法で電顕試料を得た場合)及び Fig.2 (c)、
(d) (FIB 法で電顕試料を得た場合)に示す。相対厚さから、FIB 法によって作製された試料の厚さはほ
ぼ均一になっていることがわかる。ローレンツ顕微鏡像における両矢印は試料の幅方向(W direction)
を示し、黒い矢先で示した白と黒のバンドは磁壁の位置に対応している。Fig.2(b)からは、磁区の大き
さや形が試料の形状により大きく異なっていて複雑な磁区構造をとっていることが直接見て取れる。
これに対し、FIB 法により作製された試料では磁気異方性が誘起された幅方向に平行な磁区構造をと
っていることがわかる。このことから、FIB 法を利用して形状を揃えることは磁区構造を評価するう
えで有効であることがわかる。
FIB 法によって作製された試料 A および試料 B におけるローレンツ顕微鏡像を Fig.3(a)、(b)に、矩
形の領域のホログラムをフーリエ変換することによって得た位相再生像を Fig.3(c)、(d)にそれぞれ示
す。形状が揃えられている試料 A、B のローレンツ顕微鏡像からは、観察される磁区(MD)の幅が違う
ことが分かり、磁区の幅は異方性定数(Ku)の 1/4 乗に比例することから試料の磁気異方性の違いによる
ものと考えられる 9)。ここでは試料 B の磁気異方性の方が高いため(Table.1)、観察される磁区の幅も
狭くなっている。
位相再生像 Fig.3(c)、(d)での白い点線はローレンツ顕微鏡像中で観察された磁壁に対応しており、
黒い矢印は磁束線の方向を示している。また、白い縞は実際の磁束線に対応しており、白い縞から白
い縞までの距離 d はプランク定数 h を素電荷 e で割った 4.1×10-15Wb に相当する磁束に対応する。異
方性の低い試料 A において磁束線は試料内部において閉じているが、異方性の高い試料 B においては
試料の外部に漏洩している。このことからも異方性の大きさにより磁束の分布が変化していることが
わかる。また、厚さ分布像より試料の厚さはほぼ均一であることが分かったので、試料内での内部ポ
テンシャルによる位相変化は小さく、このことからも Fig.3(c)、(d)での白い縞は基本的には磁束の特
3.
斉藤健太, 朴英吉, 村上恭和, 進藤大輔, 吉沢克仁
第 59 巻 第 1,2 号
Fig.2 Thickness distribution (a) and Lorentz microscope image (b) of a specimen Fe18.8Co60Cu0.6Nb2.6Si9B9 prepared by
ion-milling. Thickness distribution (c) and Lorentz microscope image (d) of a specimen Fe18.8Co60Cu0.6Nb2.6Si9B9
prepared by FIB. The relative thickness profiles were obtained from the rectangular regions in the insets of Fig.2 (a) and
(c).
Fig.3 Lorentz microscope images of Fe78.8Cu0.6Nb2.6Si9B9 (a) and Fe18.8Co60Cu0.6Nb2.6Si9B9 (b). Reconstructed phase
images of Fe78.8Cu0.6Nb2.6Si9B9 (c) and Fe18.8Co60Cu0.6Nb2.6Si9B9 (d).
平成 15 年 12 月
集束イオンビーム法と電子線ホログラフィーを用いた
Fe-Co-Cu-Nb-Si-B ナノ結晶磁性材料の磁区構造評価
長をよく表している。ここで、厚さ分布像から得られた試料厚さ t および磁束線の幅 d より次式を用
いて試料 A の磁束密度 B を求めた。
B=
h
d ⋅t ⋅e
(1)
その結果、1.6T という値になり、これは報告されている 1.5T というバルク試料での値とほぼ一致して
いる 3)。なお、試料 B については、Fig.3(d)からもわかるように漏洩磁場の影響が大きく、式(1)を用い
た磁束密度の評価は行なっていない。
4. 結論
(1) FIB 法によりイオンミリング法では不可能であった形状および膜厚の制御された電顕試料の作
製が可能となった。これによりローレンツ顕微鏡法および電子線ホログラフィーによる磁区構
造の評価精度が向上したと結論できる。
(2) FIB 法で得た試料によるローレンツ顕微鏡像には、磁気異方性の違いに起因する磁区構造の相異
が明瞭に示された。具体的には、磁気異方性の大きな Fe18.8Co60Cu0.6Nb2.6Si9B9 の試料の方が幅の
狭い磁区が観察された。
(3) 厚さ分布像および電子線ホログラフィーにより Fe78.8Cu0.6Nb2.6Si9B9 の試料の磁束密度は 1.6T と
見積もられた。これは報告されているバルク試料での値とほぼ一致している。
謝辞
本研究で利用した FIB 装置は、21 世紀 COE プログラム「物質創製・ 材料化研究教育拠点」のプロ
ジェクトにより導入されたものである。また本研究の遂行にあたりご支援、ご協力を頂いた早稲田嘉
夫副総長、中西八郎所長に感謝の意を表します。
文献
1)
2)
3)
4)
5)
6)
7)
8)
9)
D. Shindo; T. Oikawa: Analytical Electron Microscopy for Materials Science,(Springer-Verlag,
Tokyo,2002)
Y. Yoshizawa; S. Oguma; Y. Yamauchi: J.Appl.Phys.64(1988), 6044.
Y. Yoshizawa; S. Fuji; D.H. Ping; M. Ohnuma; K. Hono: Scripta Materialia 48(2003), 863.
G. Herzer: IEEE Trans. Magn.23(1989), 3327.
K. Hono; Y. Zhang; A. Inoue; T. Sakurai: Acta Mater. 40(1992), 2137.
D. Shindo; Y.-G. Park; Y. Yoshizawa: J.Magn.Magn.Mater. 238(2002), 101.
H.A.M. van den Berg: J.Appl.Phys.60(1986), 1104.
H.A.M. van den Berg: J.Appl.Phys.62(1987), 1952.
近角聰信 : 強磁性体の物理,(1984), 200. 裳華房
光誘導ドリフトによる同位体分離
武山昭憲*,佐藤俊一**
Isotope Enrichment of Lithium by Light-induced Drift
By Akinori TAKEYAMA and Shunichi SATO
Isotope enrichment of lithium by light-induced drift (LID) was studied. Lithium atoms collected on a Si
substrate was evaluated by SIMS. The separation factor calculated from the SIMS depth profile was 1.02.
The diffusion equation describing the distribution of lithium vapor caused by LID was solved numerically.
The concentration maps of lithium vapor indicated the isotope ratio was strongly dependent on the position of
the Si substrate.
(Received October 6th, 2003)
Keywords: laser, light induced drift, isotope separation, lithium
1.
はじめに
光誘導ドリフト( Light Induced Drift, LID )は,気体原子または分子と緩衝ガスの混合気体に,適
当な波長のレーザー光を照射した場合に生ずる物理現象である.(1)
一般に静止した原子や分子が吸収可能な光の波長は,それら自身に固有のエネルギー準位構造に
より決定される.しかし気体状の原子や分子は空間を任意に運動しているため,その運動速度に応じ
てドップラー効果が生じ,吸収可能な光の波長は各原子の速度により異なる.今,気体が原子から成
るとすると,この原子気体が吸収できる光の波長はドップラー分布とよばれる広がりを持つ.
ドップラー分布の幅より十分に狭いスペクトル幅のレーザー光を原子気体と緩衝ガスとの混合ガ
スに照射すると,特定の速度成分を持つ気体集団のみが励起される.一般に励起状態にある原子の拡
散断面積は基底状態のそれに比べて大きいため,励起状態にある原子は,緩衝ガスとの衝突により減
速されやすくなる.従って,レーザー光により特定方向の速度成分を持つ原子を選択的に励起するこ
とにより,結果的にそれと逆方向への流れが生ずる.これが光誘導ドリフトの原理である.(2)
光誘導ドリフトは原子集団が励起された結果生ずる現象であるため,レーザー光により特定の同
位体からなる原子集団のみを励起することにより,特定の同位体原子の流れを生じさせることができ
る.レーザー光により励起されない原子集団はその運動になんら変化を受けないため,結局、特定同
位体のみを移動させ濃縮することが可能となる.レーザー光による同位体分離法としては原子法、分
子法が有名であるが,同位体分離法としての光誘導ドリフトには次の優れた特徴がある.すなわち,
特殊構造のチャンバ,レーザー光は不要,系の構成要素が少ない,低コストなどが挙げられる.
光誘導ドリフトによる同位体分離は,これまでに数種類の原子および分子について試みられてい
る.Gangrsky ら(3 )は Na の放射性同位体 22, 23, 24Na の分離実験を行い,γ線の強度計測により 22Na,24Na
の分離係数α = 10~100 を得ている.また Hradecny ら(4)も同じく Na について実験を行い,分離係数と
して 25 が報告されている.Li については Atutov ら(5), Vostrikov ら(6)により同位体分離が試みられた
が,分離係数等の分離効果について定量的な評価がなされたとは言いがたい.
そこで本研究では光誘導ドリフトによる Li 同位体分離を行い,その分離効果の定量的な評価を行っ
*
東北大学大学院工学研究科
** 東北大学多元物質科学研究所
平成 15 年 12 月
光誘導ドリフトによる同位体分離
た.Li には 6Li と 7Li の2種類の安定同位体が存在するが,光誘導ドリフトにより 7Li の濃縮を行い,
質量分析により分離係数を求めた.また,光誘導ドリフトが生じている場合の Li 蒸気濃度分布につい
て定性的な計算を行い,同位体分離効果の空間依存性を調べた.
2.
実験方法
本実験で使用した実験装置は,基本的に
Diaphragm
既報(7)と同様である.チャンバはパイレッ
クスガラス製(φ50×400)であり,ステン
Si substrate
レス製アパーチャー(穴の径は 8mm)によ
り 3 つに仕切られている.チャンバ中央部
分には坩堝があり,抵抗加熱により Li 蒸気
を供給する。坩堝の下面には熱電対が接触
Crucible
しており,この温度を坩堝温度とした.坩
Luminescence
of
Li 堝温度は 543K である.レーザー光波長お
よび緩衝ガスの圧力は、既報にて光誘導ド
Fig. 1
Experimental setup.
リフトが最大になると思われる条件に設定
した.レーザー光の波長は 670.960nm に固
定し,緩衝ガスには Kr を用いてその圧力は 20Torr とした.レーザー光のスペクトル幅は 20MHz 以下
であり,アパーチャー入り口で光径が約 3mm となるように集光した.
光誘導ドリフトにより移動した 7Li を堆積させるため,Fig.1 に示すように Si 基板をチャンバ径中心
から約 8mm の位置に設置した.Si 基板上の堆積物の Li 同位体濃度を,2次イオン質量分析計(SIMS)
により測定した.1次イオン種には Cs+を用い,入射エネルギーは 2keV とした.
3.
結果と考察
図2は,Si 基板上の Li 堆積物の SIMS による深さプロファイルである.レーザー光出力は 160mW,
0.09
10000
Si
0.085
Li / 7Li
1000
7
Li
100
6
Intensity/ counts/s
28
10
1
6
Li
0
1
2
0.08
0.075
0.07
3
4
0.065
0.4 0.6 0.8
Sputter time/min
Fig. 2
Depth profile of the Li deposit by SIMS.
Laser power: 160mW.
1
1.2 1.4 1.6 1.8
Sputter time/min
Fig.3
Isotope ratio of the Li deposit.
Laser power: 160mW.
波長は 670.960nm,緩衝ガスである Kr の圧力は 2666Pa である.縦軸は2次イオン強度,横軸はスパ
ッタ時間を示す.リチウム同位体の2次イオン強度は,スパッタ開始約 20 秒後から2分間の間一定で
あり,その後,Si 強度の増加とともに減少している.基板である Si の強度はスパッタ開始後2分以降
は一定であることから,これ以降の Li 同位体の信号は基板中に拡散した Li と考えられる.従って,
武山昭憲,佐藤俊一
スパッタ開始後2分以内の深さプロファイルが,
10000
28
Intensity/ counts/s
Si
1000
7
Li
100
6
10
1
Li
0
1
2
3
4
Sputter time/min
Fig. 4
第 59 巻 第 1,2 号
Depth profile of the Li deposit by SIMS.
Laser power : 10mW.
Li 堆積物の組成を表すと思われる.
図3は,深さプロファイルから求めた Li 同位
体比 6Li / 7Li である.縦軸は同位体比,横軸は
スパッタ時間である.図中に実線および点線で
示した直線は,各々得られた同位体比の平均値
および天然同位対比を示す.天然同位体比とし
ては 6Li:7Li の存在比を 7.5:92.5 として計算し,
6
Li / 7Li =0.0811 を得た.深さプロファイルから
求めた Li 同位体比の平均値は 0.0795 である.
この値は天然同位体比よりも小さく,Li 堆積物
中に 7Li が濃縮されていることを示している.
すなわち光誘導ドリフトにより 7Li が濃縮され
た流れが生じ,Si 基板に Li 蒸気が付着したため
6
Li / 7Li
と考えられる.
SIMS の深さプロファイルの形状は,試料形態や不純物に影響を受けることが知られている.従っ
て光誘導ドリフトの同位体濃縮効果をより確かめるためには,光誘導ドリフトが生じていない状態で
実験を行い,両者の結果を比較する必要がある.図 4 は,光誘導ドリフトが生じていない状態での Li
堆積物の深さプロファイルである.レーザー光出力は 10mW,波長は 670.962nm,緩衝ガスである Kr
の圧力は 20Torr である.既報に示したように,このレーザー光の波長は 7Li の吸収スペクトルの中心
波長であること,またレーザー光出力が非常に弱いことから,この条件下では光誘導ドリフトは生じ
ないと考えられる.Li 堆積物の厚さが図 3 と異なるため,スパッタ開始から短時間において Li 同位
体の2次イオン強度は減少している.基板である Si の2次イオン強度がスパッタ開始後2分以降飽和
に近づくことから,図3と同様スパッタ時間が2分以内の Li の信号が,Li 堆積物の組成を表すと考
えられる.図 6 は,図5から求めた Li 堆積物の Li 同位体比 6Li / 7Li である.図中,実線は同位対比の
平均値,点線は天然同位対比を示す.平均値は 0.0821 であり,図からわかるように天然同位体比に非
常に近い.従って光誘導ドリフトが生じていない
0.09
状態では,たしかに Li 同位体濃縮は生じていない
0.085
と考えられる.
図4から計算した同位体分離係数は,1.02 であ
0.08
った.ただし,同位体分離係数を(同位体濃縮後
の同位体比)/(同位体濃縮前の同位体比)とし,
0.075
深さプロファイルから得られた同位対比の平均値
を天然同位体比で割った.この値は,化学交換や
0.07
イオン交換樹脂を用いた同位体分離法とほぼ同等
の値である.(8) (9) またこの値は,多元素における
0.065
0.4 0.6 0.8 1 1.2 1.4 1.6 1.8 光誘導ドリフトを利用した同位体分離法と比べて
も非常に小さい.このように同位体濃縮効果が小
Sputter time/min
さい原因として,元素の種類およびレーザー光に
Fig.5 Isotope ratio of the Li deposit.
起因する本質的なもの,チャンバ中の流れや Li
Laser power: 10mW.
蒸気濃度分布に起因するものの二つが考えられる.
いずれを考える上でも,本実験と Rb の光誘導ドリフトの実験との対比は非常に有効である.特に本
研究室で行われた Rb の光誘導ドリフトの実験は,チャンバ形状が同様であることに加え実験条件が
明確であり,比較対象として最適と思われる.(10)
光誘導ドリフトの効果の大きさを表す代表的なパラメータは,基底状態,励起状態にある原子間の
平成 15 年 12 月
光誘導ドリフトによる同位体分離
拡散断面積の差である.Hamel ら(11) による拡散断面積から計算すると,基底状態と励起状態原子間の
規格化拡散断面積の差は,緩衝ガスとして Kr を用いた場合 Li,Rb 各々29.885, 45.247 であり,Rb
の方が大きい.このことは,励起された Rb 原子の方が Kr 原子との衝突により減速されやすいことを
示している.しかし光誘導ドリフトの効果は,ドップラー幅およびレーザー光のスペクトル幅にも依
存する.Na は光誘導ドリフトの効果が大きいことが知られているが,Li においてもスペクトル幅が
広いレーザー光を用いた場合,Na に匹敵するドリフト速度が報告されている.すなわち,ドップラー
幅に対してある程度広いスペクトル幅を持つレーザー光が有効と推測される.
各々の実験条件においてドップラー幅を計算すると,Li では 2.8GHz,Rb では 550MHz である.い
ずれの実験においてもレーザー光のスペクトル幅は 40MHz 以下であるが,Li のドップラー幅は Rb の
5倍強であるため,レーザー光により励起される原子数の全原子数に対する割合は,Li の方が小さい.
従って,本実験で用いたレーザー光のスペクトル幅程度では,十分な Li の光誘導ドリフトの効果を得
ることは難しいと推測される.
同位体濃縮効果が小さい理由として,チャンバ中の流れおよび蒸気濃度分布の影響が考えられる.
まず,光誘導ドリフトの効果を阻害する流れとしては,試料蒸気の自然対流が考えられる.Li は Rb
よりも軽く,また十分な蒸気を発生させるためにはより高い温度が必要なため,チャンバ中の対流が
より起きやすい.
本実験のように,チャンバ中に設置した基板に試料蒸気を堆積させて回収を行う場合,堆積物の同
位体濃度は基板位置に強く依存する.したがって同位体濃縮度を正確に把握するためには,チャンバ
中の Li 蒸気の分布を知ることが非常に有効である.本実験のような管状チャンバ中における Li 蒸気
濃度分布は,以下の移流―拡散方程式に従うと考えられる.(12)
∂N / ∂ t = D ( ∂ 2 N / ∂ r 2 + 1 / r ∂ N / ∂ r + ∂ 2 N / ∂ r 2 ) − u ∂ N / ∂ z LL (1)
N:リチウム蒸気濃度
D:リチウム蒸気の拡散係数 u:ドリフト速度
正確な Li 蒸気濃度分布を求めるためには,(1)式においてドリフト速度の空間的な分布を考慮する必
要があるが,ドリフト速度の分布を無視した場合でも定性的には分布形状は変わらない.(12) そこで,
本実験系を十分定性的に表すと思われる移流―拡散方程式について,いくつかのドリフト速度につい
て Li 蒸気濃度を求めた.
∂N / ∂ t = D ∂ 2 N / ∂ r 2 − u ∂ N / ∂ z LL (2)
N = 1,
r=R
r ≦ a,
N = 0,
at z > 0
a : アパーチャー半径(mm)
r ≻ a
a = 4,
at z = 0
R = 25
R : チャンバ半径(mm)
実験系は定常状態と考え,左辺を 0 とすることにより定常解が得られる.アパーチャー開口部ではリ
チウム蒸気濃度分布は無視できるとして,この蒸気濃度を1とした.開口部以外のチャンバ壁面にお
いてリチウム蒸気は壁面に速やかに吸着されると考えられ,リチウム蒸気濃度はチャンバ壁面におい
て0とする.リチウム蒸気の Ar 中での拡散係数は,文献値(13)を使用した.
Fig. 6,Fig.7,Fig 8 はいくつかのドリフト速度について計算した Li 濃度分布である.
各々,u/D=1000,
500,200 に対応しており,縦軸はアパーチャーからの距離,横軸はアパーチャー中心からの距離を表
す.また,色が薄い部分ほど Li 蒸気濃度は高い.式(2)は Li 蒸気のレーザー光軸方向およびレーザー
光径方向の拡散項を無視しているため,Li 蒸気の分布は報告されている値よりも広がるが,同様の形
状が得られる.図よりドリフト速度が大きくなるにつれて,レーザ光径方向の分布は小さくなり,レ
ーザー光軸方向の濃度分布が大きくなることがわかる.
第 59 巻 第 1,2 号
武山昭憲,佐藤俊一
40
Axial Distance / mm
Axial Distance / mm
40
30
20
10
0
Fig. 6
0
5
10
15
Radial Distance / mm
0
Fig. 7
5
10
15
Radial Distance / mm
20
Contour maps of lithium vapor
concentration. u /D = 500.
40
Axial Distance / mm
Axial Distance / mm
10
0
Contour maps of lithium vapor
concentration. u /D = 1000.
30
20
10
Fig. 8
20
20
40
0
30
0
5
10
15
Radial Distance / mm
30
20
10
0
20
Contour maps of lithium vapor
concentration. u /D = 200.
0.05
0
Fig. 9
5
10
15
Radial Distance / mm
20
Contour maps of the isotope ratio,
6
Li/ 7Li. u/D of 6Li = 800.
u/D of 7Li = 1000.
光誘導ドリフトはレーザー光により励起された原子のみに生ずるため,7Li の濃度分布はドリフト
速度に応じて Fig.6,Fig.7,Fig.8 の濃度分布に従うと思われる.したがって,チャンバ内に光誘導ド
リフト以外の流れが存在せず,また 6Li が光誘導ドリフトにより生ずる流れの影響を全く受けない場
合,Fig.6,Fig.7,Fig.8 は 7Li の濃縮度を表す.
しかし実際には,レーザー光を吸収しない 6Li や不純物原子は,チャンバ内の自然対流や 7Li の光
誘導ドリフトに巻き込まれて移動すると考えられる.特に本実験のように光誘導ドリフトの効果が小
さい場合 6Li と 7Li の流速はほぼ等しく,濃度分布はほぼ等しい形状になると予想される.そこで,6Li
の流速が 7Li の 80%程度であると仮定して 6Li の濃度分布を計算し,同位体比 6Li/ 7Li の空間分布を求
めた.なお,式(2)の計算にあたって光誘導ドリフトの効果は小さいと仮定し,アパーチャー付近にお
ける 7Li,6Li の比は天然同位体比に等しいとした.すなわち,
7
Li : N = 0.925, r ≦ a,
6
Li : N = 0.075, r ≦ a,
7
6
Fig.9 は, Li の u/D = 1000, Li の u/D = 800 とした場合の同位体比 6Li/ 7Li である.図中の数字は同位
平成 15 年 12 月
光誘導ドリフトによる同位体分離
対比を示す等高線を表しており,線の左側の部分における同位対比の最低値を示す.同位体比が 0.05
の場合 6Li: 7Li = 4.8 : 95.2 であり,アパーチャー中心から 10mm 以内の範囲では 7Li が 95%以上に濃縮
されることがわかる.レーザー光軸方向についてみると,アパーチャーから離れるにしたがって同位
体比が小さい領域が広がっていることがわかる.これは 6Li の流速が遅いために,相対的に 6Li の濃度
がアパーチャー付近で高くなるためである.一方レーザー光径方向については,アパーチャー中心か
ら遠ざかるほど,同位体比は大きくなっている.これは,チャンバ壁面に近づくにつれて Li 蒸気濃度
が低くなり,6Li と 7Li の濃度差が小さくなるためと思われる.従って、この領域に基板を設置した場
合,見かけ上,高濃度の 6Li を採取することが可能であるが,その量は極めて少ないと予想される.
本計算では,光誘導ドリフトの効果が小さく 6Li と 7Li の流速の差は小さいとしたが、ドリフト速度が
大きくなるにつれて,同位体比が小さい領域が広がると考えられる.式(2)はレーザー光軸,および径
方向の Li 蒸気の拡散を無視しており,またドリフト速度の空間分布を考慮していないため,実際の同
位対比の分布はより狭まると考えられる.また,光誘導ドリフトによる同位体分離効果を議論する場
合には,濃度分布を積分し,全蒸気濃度について考慮する必要がある.しかし本計算により光誘導ド
リフトが生じている場合,基板堆積物の同位体濃度は,基板位置に強く依存することが定性的に示さ
れた.
4.
おわりに
光誘導ドリフトにより,Li 同位体 7Li を濃縮した.基板上堆積物の 2 次イオン質量分析計による深
さプロファイルから求めた同位体分離係数は,1.02 であった.移流−拡散方程式を解くことにより,
光誘導ドリフトが生じている場合の Li 同位体の定性的な濃度分布を求め、堆積物の同位体分離効果は
基板位置に強く依存することを示した.
文献
(1) F. Kh. Gel’mukhanov and A. M. Shalagin: JETP Lett., 29(1979), 711.
(2) G. Nienhuis: Phys. Rep., 138(1986), 151.
(3) Yu. P. Gangrsky, C. Hradecny, J. Slovak, T. Tethal, I. M. Yermolayev : Phys. Lett. A, 168(1992), 230.
(4) C. Hradecny, T. Tethal, I. M. Yermolayev, S. G. Zemlyanoi, P. Zuzaan : Appl. Radiat. Isot., 45(1994), 257.
(5) S. N. Atutov, P. V. Kolinko, A. M. Shalagin : Laser Phys., 3(1993), 855.
(6) O. A. Vostrikov, K. A. Nasyrov, S. P. Pod’yachev, A. M. Shalagin : JETP, 87(1998), 634.
(7) A. Takeyama, S. Sato : Mater. Trans. JIM, 41(2000), 1108.
(8) K. Nishizawa, H. Watanabe, S. Ishino and M. Shinagawa : J. Nucl. Sci. Technol., 21(1984), 133.
(9) H. Takahashi and T. Oi : J. Mater. Sci., 36(2001), 1621.
(10) S. Sato, M. Saito : Mater. Trans. JIM, 37(1996), 1789.
(11) W. A. Hamel, J. E. M. Haverkort, H. G. C. Werij, J. P. Woerdman : J. Phys. B, 19(1986), 4127.
(12) S. N. Atutov, P. V. Kolinko, A. M. Shalagin, O. A. Vostrikov : Opt. Comm., 128(1996), 236.
(13) S. N. Atutov, B. V. Bondarev, S. M. Kobtzev, P. V. Kolinko, S. P. Podjachev, A. M. Shalagin : Opt. Comm.,
115(1995), 276.
液相還元選択析出法による Ni-Zn ナノ粒子の成長機構と物性
サラントヤミャグマルジャブ*,高橋英志*,砂川洋二*,
山本勝俊*,佐藤修彰*,村松淳司*
Growth Mechanism and Characterization of the Ni-Zn Nanoparticles on TiO2 Fine Particles
Synthesized by the Liquid Phase Reductive Deposition Method
By Sarantuya MYAGMARJAV, Hideyuki TAKAHASHI, Yoji SUNAGAWA,
Katsutoshi YAMAMOTO, Nobuaki SATO and Atsushi MURAMATSU
The growth mechanism of Ni-Zn/TiO2 nanocomposite synthesized by the liquid phase reductive
deposition method and characterization were studied. Nanoparticles grew at the surface of TiO2, and the
number of growth site of nanoparticles increased with increasing the Zn concentration. Zn complex remained
in the solution adsorbed at the surface of nascent state nanoparticles and catalytically reacted, and Zn layer at
the surface of the nanoparticles also restricted the growth of nanoparticles. Thus, the size of nanoparticles
was decreased with increasing the amount of Zn added. The state of Ni in the nanoparticles was metal, and
the state of Zn and B were considered to oxide.
(Received October 17th, 2003)
Keywords: Growth mechanism, Ni-Zn nanoparticle, Selective deposition, Liquid-phase reduction method,
1.緒論
物質の粒子径をナノメートルオーダーまで減少させると電子状態などの物性が変化し (量子サイ
ズ効果 ), 同一の物質であってもサイズごとに異なった物性を示すことや 1), 単位重量あたりの表面
積増加に伴い触媒活性向上が期待できるなど 2),次世代の材料としてナノ粒子は大きな注目を集め
3-5)
ている.この様なナノ粒子の合成法には様々な手法が報告されているが
,気相反応で 2 種類以上
の金属を含有する金属状態のナノ粒子を製造することは,それぞれの金属の蒸気圧が相違するなど
の問題から極めて難しい 6).それに対し,我々が開発した液相還元法は大掛かりな装置を必要とせず
実験室規模で容易にほぼ 100%の回収率で金属ナノ粒子を合成できる手法である.本方法は溶液に可
溶な金属錯体を用いれば, 様々な金属への応用や2種以上の金属の複合化も可能であることから, 有
効な金属ナノ粒子合成手段の一つである.しかしながら, 液相還元法により調製した金属ナノ粒子は
その高い表面活性のために反応中に凝集・ 凝結して失活することが多い.この様な好ましくない副反
応を抑制するため, 酸化チタンナノ粒子表面上へ金属ナノ粒子を選択析出する手法を開発し, 酸化チ
タンが凝集の抑制と合成された金属ナノ粒子の安定化に効果を示すこと, Ni ナノ粒子合成時に Zn を共
存させると粒子径の減少と水素化触媒活性に効果を示すことを報告してきた 7,8).
本研究では, これまで明らかにされていない液相還元選択析出法による Ni-Zn ナノ粒子の成長機構
と物性について検討を行った.
* 東北大学多元物質科学研究所
平成 15 年 12 月
液相還元選択析出法による Ni-Zn ナノ粒子の成長機構と物性
2.実験
金属ナノ粒子の合成は以下の手順にて行った.ニッケルアセチルアセトナート(以下 Ni(AA)2)及び
亜鉛アセチルアセトナート(以下 Zn(AA)2)の 2-プロパノール溶液 40ml(Zn/Ni=0〜1.0, [Ni]= 2.5x10-4mol
一定)及び 125mg の酸化チタンナノ粒子(ST01 粒子, 石原産業製, 表面積:約 300cm2/g, 以下 TiO2)を
4 つ口フラスコ中で 30 分の超音波照射を行うことにより分散させ, Ni 及び Zn 出発物質が吸着平衡に
この状態で ST01 粒子は高分散状態を維持しており, 沈殿は起こさない.
至るまで更に 30 分放置する.
還 流 管 を 取 り 付 け 窒 素 ガ ス 気 流 中 で 所 定 温 度 (-10 ℃ 〜 82 ℃ ) に 制 御 し 溶 存 気 体 の 置 換 後 ,
1.0x10-3mol·dm-3 水素化ホウ素ナトリウム/2-プロパノール溶液 10ml を滴下することで液相還元を開始
する.所定時間(1 分〜180 分)の加熱・ 攪拌後, 試料分散液 1ml を取り出し-10℃に冷却した 2-プロパノ
ール 9ml 中に投入することで還元反応を停止させる.この分散液をメンブレンフィルター (ポア径
0.1µm)で濾過し, 濾液中の Ni 及び Zn 濃度は誘導結合型プラズマ発光分析 (島津製作所製 ICPS-1000Ⅲ,
Inductively Coupled Plasma Atomic Emission Spectrometer,以下 ICP 分析,λNi=221.647nm,λZn=213.856nm,
ニッケル標準液(Ni(NO3)2 in 0.1mol/l・ HNO3,和光純薬製)
,亜鉛標準液(Zn(NO3)2 in 0.1mol/l・ HNO3,
和光純薬製))を用いて, 合成された Ni-Zn/TiO2 ナノコンポジットは高分解能 FT-IR(デジラボジャパン
製,FTS-7000 システム, 0.5wt% KBr ペレット 100mg), 透過型電子顕微鏡(日立製作所製 電界放射型
HF-2000,加速電圧:200KV,エミッション電流:約 20µA,以下 HR-TEM),X 線光電子分析装置(Electron
Spectroscopy for Chemical Analysis,多元物質科学研究所多機能型素材分析装置 ULVAC-PHI ESCA 5600,
Al Kα,照射電子:187.85eV,照射角:45°,以下 ESCA)及びX線吸収微細構造装置(Extended X-ray
Absorption Fine Structure, 理学社製 R-XAS Looper, 以下 EXAFS)を用いて評価した.ESCA 測定試料は
十分な洗浄後に得られたナノ粒子を空気に触れることがないよう 2-プロパノール中で保管し,試料
台へはアルゴンガスで置換したグローブボックス中で滴下および乾燥を行い測定した.EXAFS 測定
試料はナノ粒子合成後,遠心分離により濃縮し容器中に密封して測定に用いた.
また, 1x10-6 〜1x10-1 mol·dm-3 の Ni(AA)2 単独, Zn(AA)2 単独及び Ni(AA)2/Zn(AA)2 混合溶液中に
100mg の ST01 粒子を超音波分散し, 25℃のオイルバス中で 24 時間保持し, 濾液中の Ni 及び Zn 濃度
を ICP で分析することにより吸着等温線を得た.
3.結果および考察
3.1 Ni-Zn/TiO2 ナノコンポジットの成長機構
TiO2 表面上への Ni(AA)2 及び Zn(AA)2 の吸着等温線は Langmuir 式に従った.表1は吸着等温線から
得られた Ni(AA)2 及び Zn(AA)2 の飽和吸着量を示す.Zn の飽和吸着量は Ni の有無に関わらず 2.8mmol/g
であったのに対し, Ni の飽和吸着量は Ni 単独の場合は 1.3mmol/g, Zn/Ni=1.0 の場合 2.0mmol/g となり,
Zn 添加により Ni 吸着量は増加した.Ni の飽和吸着量が Zn の添加により増加することは, 酸化チタ
ン表面上でのナノ粒子生成サイトの増加を意味すると考えられ, その結果として生成するナノ粒子の
径が減少すると考えられる.
Ni alone
Zn alone
Zn/Ni=1.0
Table1 Saturated adsorption amount of Ni(AA)2 and Zn(AA)2 onto TiO2.
Saturated adsorption amount of Ni
Saturated adsorption amount of Zn
1.3mmol/g
2.8mmol/g
2.0mmol/g
2.8mmol/g
Fig.1 は還元剤投入前と投入後 1, 10 及び 30 分経過後の Ni-Zn/TiO2 ナノコンポジット(Zn/Ni=0 及び
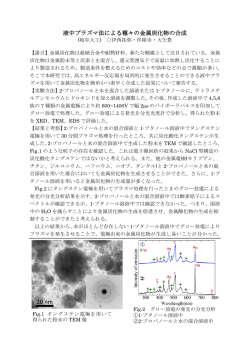
0.5,82℃で合成)の HR-TEM 像を示す.いずれの場合も還元剤投入前は担体の TiO2 のみ観察される.
(a)Zn/Ni=0 の場合,還元剤投入 1 分後には 1nm 程度のナノ粒子が生成しており, 反応時間の経過と共
サラントヤミャグマルジャブ, 高橋英志, 砂川洋二, 山本勝俊,
佐藤修彰, 村松淳司
第 59 巻 第 1,2 号
に粒子は成長し還元剤投入 30 分後には 5-6nm 程度に成長することが観察できる.それに対し ,
(b)Zn/Ni=0.5 の場合は還元剤投入 1 分後に 1nm 以下のナノ粒子が生成しているが,還元剤投入 30 分後
でも粒子径は 1nm 程度であることがわかる.Zn/Ni=1.0 の場合の HR-TEM 像は Zn/Ni=0.5 の場合とほ
ぼ同様であった.以上の結果から,液相還元選択析出法によるナノ粒子合成では,溶液中でナノ粒子
が生成し表面に付着するのではなく,最初に TiO2 表面上に小さい核が生成し時間の経過と共に粒子が
成長すること,Zn の添加によりナノ粒子の成長が抑制されることがわかる.
Fig.1 HR-TEM micrographs of Ni-Zn/TiO2 nanocomposites synthesized at the condition of (a) Zn/Ni=0 and
(b) Zn/Ni=0.5, before and after reduction (1, 10 and 30min).
Fig.2 FT-IR spectra of Ni-Zn/TiO2 nanocomposites synthesized at the condition of Zn/Ni=0, 0.3 and 0.5,
before and after reduction (1, 5, 10, 20 and 30min), and the reduction rate of Ni-Zn/TiO2 nanocomposites
calculated from the FT-IR results.
Fig.2 は還元剤投入前と投入後 1, 5, 10, 20 及び 30 分経過後の Ni-Zn/TiO2 ナノコンポジット(Zn/Ni=0, 0.3
及び 0.5,70℃で合成)の高分解能 FT-IR 測定結果とピーク面積より計算された還元反応速度を示す.
平成 15 年 12 月
液相還元選択析出法による Ni-Zn ナノ粒子の成長機構と物性
還元反応速度は以下の手順にて算出した.0.1-0.5wt%の Ni(AA)2 及び Zn(AA)2 の KBr ペレット 100mg
の FT-IR 測定結果から得られたピーク面積(1262cm-1 及び 1517cm-1)を用いて作成した検量線から試料
中に含まれる Ni(AA)2 及び Zn(AA)2 の量を算出した.検量線は含有量の増加と共に直線的に増加し,
(1262cm-1 のピーク面積)/(1517cm-1 のピーク面積)が一定となることから,いずれのピークを用い
ても検量線から試料中の含有量は算出できる.本実験においては 1262cm-1 のピーク面積から含有量を
算出した.還元剤投入 1 分後の含有量を初期濃度(A0),各時間における含有量を A,k を反応速度定数
とする.反応速度式–d[A]/dt=k[A]を積分することにより log([A0]/[A])=kt/2.303 が得られる.反応時間 t
を横軸,log([A0]/[A])を縦軸にとれば直線の勾配が k/2.303 となり,勾配が大きいほど反応速度定数が
大きい(還元反応速度が早い)ことになる.
高分解能 FT-IR 測定結果から,還元剤投入前はいずれの条件の場合でも小さくブロードなピークの
みが観測される.還元剤投入前の試料中には Ni(AA)2 及び Zn(AA)2 が吸着した酸化チタンが 0.5wt%の
濃度で含有されているが,吸着量より算出される試料中の Ni(AA)2 及び Zn(AA)2 の含有量は検出限界
以下であることがわかった.したがって還元剤投入前の試料にもこれらの種は存在するが,ピークと
しては検出されなかったと考えられる.還元剤投入 1 分後, AA の存在を示す多数のピーク(図中●)が観
測されるようになり, Zn/Ni=0 及び 0.3 の場合は還元剤投入 10 分後以降で,Zn/Ni=0.5 の場合は還元剤
投入 30 分後にそれらのピークは観測されなくなる.還元剤投入により AA のピークが観測されたこと
は溶液中の Ni(AA)2 及び Zn(AA)2 が酸化チタン表面上に吸着したこと,時間の経過によりそれらのピ
ークが消滅することは Ni(AA)2 及び Zn(AA)2 が還元され試料中から AA 基が離脱したことを示す.反
応後溶液の ICP 分析の結果から,還元剤投入 1 分後には溶液中の Ni 及び Zn はほとんど残留していな
いことがわかった.以上の結果から,Ni(AA)2 と Zn(AA)2 は還元剤投入後の初期段階において TiO2 表
面上に全て吸着し, 表面上において還元され Ni-Zn ナノ粒子が生成すると共に配位子(AA)は TiO2 表面
上から脱離することがわかる.ピーク面積より計算された還元反応速度は Zn/Ni=0 の場合に最も早く,
Zn/Ni=0〜0.3 では Zn/Ni=0 の反応速度に近い値となり,Zn/Ni>0.3 で急激に遅くなることがわかる.
Fig.3 FT-IR spectra of Zn(AA)2 adsorbed (a) TiO2, (b) TiO2 treated with NaBH4 and (c) TiO2 supported with
Ni nanoparticles.
Fig.3 は (a)TiO2, (b)NaBH4 で処理を行った TiO2 及び (c)Ni ナノ粒子を担持した TiO2,それぞれの分
サラントヤミャグマルジャブ, 高橋英志, 砂川洋二, 山本勝俊,
佐藤修彰, 村松淳司
第 59 巻 第 1,2 号
散液中に Zn(AA)2 溶液(Zn/Ni=0.5 に相当する Zn(AA)2 量)を投入した試料の高分解能 FT-IR 測定結果
を示す.図から,Ni ナノ粒子を担持した TiO2 表面上への Zn(AA)2 の吸着量が多いことがわかる.こ
の分散液中に還元剤を投入したところ, Zn(AA)2 のピーク強度は測定限界以下となった.以上の結
果から溶液中に存在する Zn(AA)2 は Ni 表面上に吸着しその表面上で触媒反応的に還元されるが,
Zn(AA)2 単独では還元されないことがわかる.従って,本手法におけるナノ粒子合成系では,還元
剤投入前に TiO2 表面上に吸着した Zn(AA)2 は , 還元剤投入により生成した Ni ナノ粒子が成長し , そ
の表面が吸着 Zn(AA)2 近傍に到達すると還元されると考えられ,還元剤投入前に吸着せずに溶液中
に残存した Zn(AA)2 は還元剤投入により生成した Ni ナノ粒子表面上に吸着し還元されると考えられ
る. Zn 添加量が多い場合,前述の飽和吸着量増加 (ナノ粒子生成サイト数の増加 )による粒子径減少
の効果と共に,溶液中に増加した遊離の Zn(AA)2 が,還元剤投入により生成した小さい Ni ナノ粒子
表面に選択的に吸着及び還元されるが更なる成長を抑制する (Zn 表面上では Zn は還元されない ,ま
た還元剤投入直後に溶液中の全ての Ni(AA)2 は試料上に吸着しているため Zn 表面上に新たに溶液中
から Ni(AA)2 が供給されないため .)ことも , Zn 添加によってナノ粒子径が減少する原因となっている
と考えられる.
3.2 Ni-Zn/TiO2 ナノコンポジット中の Ni, Zn 及び B の状態分析
3.2.1 Ni の状態分析
Fig.4 ESCA spectra for the Ni 2p3/2 region of before and after etching the samples and EXAFS results at Ni
k-edge of the nanoparticles prepared at Zn/Ni=0, 0.5 and 1.0.
Fig.4 は調製した Ni-Zn/TiO2 ナノコンポジット中の Ni の 2P3/2 軌道電子の結合エネルギーの ESCA に
よる測定結果(上段)と Ni k-edge での EXAFS による解析結果(下段)を示す.ESCA による状態分
析結果では,いずれの条件で合成したナノ粒子の場合でも,ピーク強度に差はあるが,as-prepared 試
料からは Ni 金属(852.5eV)と Ni 酸化物(Ⅱ価,855eV)のピークが観測され 9)-11), 3kV で 10 分間のス
平成 15 年 12 月
液相還元選択析出法による Ni-Zn ナノ粒子の成長機構と物性
パッタリングを行った試料では酸化物由来のピークが消滅する.この結果から,いずれの場合も試料
作成時に試料表面に不純物(酸素)が沈着しているが,ナノ粒子内部の Ni は金属状態であることが
わかる. EXAFS による解析結果(点線:測定結果,実線:計算結果)から,Zn/Ni=0 の場合は Ni の
第一近接には酸素と Ni が存在するが Ni-O-Ni や Ni-Ni-Ni 等の第二近接に関する明瞭なピークは観測
されないこと,Zn/Ni=0.5 及び 1.0 の場合は Ni の第一近接として酸素のみが明瞭なピークとして観測
されることがわかる.
いずれの場合でも Ni -Zn 及び Ni -B に関する明瞭なピークは観測できなかった.
本手法で作成されるナノ粒子はアモルファス的な構造を有することを考慮すると12),Zn/Ni=0 の場合
は粒子径が 5-6nm と他の合成条件より大きいため粒子内部に存在する短範囲の規則的配列(Ni アモル
ファスナノ粒子中の Ni-Ni 間の結合)が観測されたと考えることができ,Ni は金属の状態であり表面
は酸化されたと考えられる.Zn/Ni=0.5 及び 1.0 の場合,粒子径が 1nm 程度と小さく,粒子を構成する
Ni の数に対する表面の Ni の数が多いため EXAFS 測定結果では短範囲の規則的な配列よりも表面に存
在する Ni の状態が明瞭に観察されたと考えられるが,ESCA 測定結果から Zn/Ni=0 の場合と同様に金
属状態の Ni が存在することが明らかであるため,この場合もナノ粒子内部の Ni は金属状態であると
考えられる.
3.2.2
Zn の状態分析
Fig.5 は調製した Ni-Zn/TiO2 ナノコンポジット(Zn/Ni=1.0)中の Zn の 2P3/2 軌道電子の結合エネル
ギーの ESCA による測定結果(左)と Zn k-edge での EXAFS による解析結果(右)を示す.ESCA に
よる Zn の状態分析結果からは 1 本のピークのみが観測されるが,図中に同時に示すように金属と酸
化物(II 価)のピーク位置が近い位置に存在するため同定は困難であった.EXAFS による測定結果から
は Zn の第一近接には酸素が存在するが Ni の場合と同様に第二近接に関するピークや Zn - Ni 及び Zn
-B に関するピークは観測されなかった.オージェ電子測定結果から Zn の 2P3/2 軌道電子の結合エネル
ギーとオージェ電子の運動エネルギーの交点が酸化物領域に観測されること 13),Zn は難還元性である
ことを考慮すると,本研究で作成した試料中の Zn は酸化物の状態であると推察される.
Fig.5 ESCA spectrum for the Zn 2p3/2 region and EXAFS results at Zn k-edge of the nanoparticles prepared
at Zn/Ni=1.0.
3.2.3 B の状態分析
本研究で作製した Ni-Zn/TiO2 ナノコンポジットの元素分析を行うと,
試料中の B 含有量は 26.7%(Zn
無添加)及び 39.4%(Zn/Ni=1.0)と,Zn の添加により増加する傾向があることが明らかとなった.Ni は B
とアモルファスを形成し 2 元状態図からも金属間化合物が存在するのに対し,Zn は B とは金属間化
合物や固溶体も形成しないことが知られている.従って,Zn 含有量の増加に伴って試料中の B 含有
量が増加することは矛盾する.そこでその原因を検討するため B の状態分析を行った.
Fig.6 は調製した Ni-Zn/TiO2 ナノコンポジット(Zn/Ni=0.5)中の B の 1s 軌道電子の結合エネルギーの
サラントヤミャグマルジャブ, 高橋英志, 砂川洋二, 山本勝俊,
佐藤修彰, 村松淳司
第 59 巻 第 1,2 号
B の状態分析結果からは酸化物の明瞭なピークが観測されることから,
ESCA による測定結果を示す.
還元剤由来の B は副生産物として B2O3 の状態で試料中に存在すると考えられる.Zn 添加量の増加に
より, 還元反応に要する NaBH4 量も増加するため, 試料中の B 量が増加したと推察される.
Fig.7 ESCA spectrum for the B 1s region of the nanoparticles prepared at Zn/Ni=0.5.
以上の結果より , 本研究で作製された Ni-Zn/TiO2 ナノコンポジット中のニッケルは表面が酸化され
た金属状態であり, 亜鉛及びホウ素は酸化物の状態で存在すると考えられる.
4.結論
本研究では液相還元選択析出法による Ni-Zn/TiO2 ナノコンポジットの成長機構と生成したナノ粒子
の状態分析を行った.成長機構は以下の通りである.
(1)TiO2 担体表面上にナノ粒子の生成サイトとなる Ni(AA)2 が吸着する. Zn(AA)2 を添加すると
Ni(AA)2 の吸着量(=ナノ粒子の生成サイト数)が増加し,粒子径が減少する原因となる.
溶液中の遊離の Ni(AA)2 が析出・ 還元され TiO2
(2)還元剤を投入すると小さいナノ粒子の核が生成し,
表面上でナノ粒子は成長する.
(3)Zn(AA)2 の添加量を増加すると遊離の Zn(AA)2 量が増加し,それらは還元剤投入により発生した
微小な Ni ナノ粒子表面上に析出し,Ni 表面でのみ触媒反応的に還元される.
(4)ナノ粒子表面で還元析出した Zn 表面では Zn(AA)2 は還元されず,新たに Ni(AA)2 が溶液中から
供給されないこともナノ粒子の成長を抑制する原因となっている.
生成した試料中に含まれる Ni, Zn 及び B の状態は以下の通りである.
(5)Ni は金属の状態で存在する.
(6)Zn 及び B は酸化物の状態で存在する.
謝辞
XPS 分析及び FT-IR 分析に多大なご協力を頂いた東北大学多元物質科学研究所技術部 佐藤史生
技官および伊東益雄技官,東北大学多元物質科学研究所 多元ナノ材料研究センターに深く感謝い
たします.
文献
1)R.Kubo, J.Phys. Soc.Jpn., 17, 975, (1962)
2)触媒学会編 ,"触媒講座 第 5 巻 触媒設計", 講談社サイエンティフィック , 84, (1985)
平成 15 年 12 月
液相還元選択析出法による Ni-Zn ナノ粒子の成長機構と物性
3)P. N. Barnes, P. T. Murray, T. Haugan, R. Rogow and G. P. Perram: Physical C-Superconductivity and its
Applications 377 (2002) 578-584.
4)K. Wegner, B. Walker, S. Tsantilis and S. E. Pratsinis: Chemical Engineering Science 57 (2002) 1753-1762.
5)B. Xia, K. Okuyama and I. W. Lenggoro: Advanced Materials 13 (2001) 1744-1744.
6)"触媒科学", 丸善,御園生誠 ,斉藤泰和共著, 71, (1999)
7)村松淳司 , 設楽修一,佐々木弘,臼井進之助 : 資源と素材 , 106, 799-804, (1990)
8)高橋英志,村松淳司, 松原英一郎, 早稲田嘉夫: 資源と素材, 118 (2002) 211-216.
9)A.Lebugle, U.Axelsson, R.Nyholm, N.Martensson, Phys, Scr., 23, 825, (1981)
10)T.Dickinson, A.F.Povey, P.M.A.Sherwood, J.Chem. Soc. Faraday Trans., 173, 332, (1977)
11)K.Ng, D.M.Hercules, J.Phys. Chem., 80, 2095 (1976)
12)H.Takahashi, Y.Sunagawa, S. Myagmarjav, K.Yamamoto, N.Sato, A.Muramatsu, Mat. Trans., 44, 11, (2003)
2414-2416.
13)砂川洋二,高橋英志,サラントヤミャグマルジャブ,山本勝俊,佐藤修彰,村松淳司 :
東北大学多元物質科学研究所素材工学研究彙報 5, 37-44 (2002)
希土類−シリコン−酸窒化物
RE4Si2O7N2(RE-J-phase,
RE =希土類)の結晶構造
高橋純一*,山根久典**,島田昌彦*
Crystal Structure of Rare-earth Silicon-oxynitrides RE4Si2O7N2
(RE-J-phase, RE = rare earth)
By Junichi TAKAHASHI, Hisanori YAMANE and Masahiko SHIMADA
1. はじめに
高温構造材料である Si3N4, AlN, SiC などの非酸化物系セラミックスの分野においては,粒界相制御
による高温機械的特性の改善が盛んに試みられている.高温における機械的特性の低下は融点の低い
SiO2 を多く含むガラス相に起因するため,高融点の結晶性粒界相を形成させ,かつ,高密度の焼結体
を得るため,MgO,BeO,希土類酸化物などの酸化物が焼結助剤として添加されている 1).また,Al2O3
を添加した系は SiAlON と呼ばれる酸窒化物群を形成することでよく知られている.近年の Si3N4 焼結
体における研究では,母材の特性を極力生かしつつ材料特性を改善するため,焼結助剤量を減らす努
力がなされている.最近,Yb2O32)や Lu2O33)を焼結助剤として数 wt%添加した Si3N4 焼結体が,他の希
土類酸化物添加の焼結体に比べて優れた高温機械強度を有することが明らかにされた.これらの焼結
体では粒界に Yb4Si2O7N2 や Lu4Si2O7N2 など,RE-J-phase(RE = 希土類)と呼ばれる希土類−シリコ
ン−酸窒化物が観察されている. Si3N4-SiO2-Yb2O3 系 4),および,Si3N4-SiO2-Lu2O3 系状態図 5)の研究か
ら, Yb や Lu などの重希土類を含む系では微量の希土類酸化物添加で粒界部に耐酸化性が低い
RESiO2N やメリライト(RE2Si3O3N4)相を存在させず,高融点の RE-J-phase を生成させることが可能
であることが明らかにされている.
Si3N4 焼結体の研究とともに,1970 年代中期から 1990 年代終わりにかけて RE-J-phase の単相試料が
合成された.RE-J-phase が生成しない Pm と Eu を除く RE = Y,および,La-Lu の J-phase について,
X線回折データ,および,格子定数が報告された 4),6)-19).これらの研究では,RE-J-phase の結晶構造は
cuspidine (Ca4Si2O7F2,単斜晶系空間群 P21/c)型と考えられ,X線回折図形の指数付けが行われた.最
近,筆者らは La-J-phase(La4Si2O7N2)20),および,Lu-J-phase(Lu4Si2O7N2)21,22)の単一相を合成し,
X線および中性子粉末回折データを用いたリートベルト(Rietveld)法により格子定数や原子座標,窒
素と酸素の占有率などの構造パラメータを精密化した.その結果, Lu-J-phase は従来言われていた
cuspidine 構造であるが,La-J-phase は cuspidine 構造とは b 軸方向の原子の積み重なり方が異なる類型
構造であることが明らかとなった 20).また,精密化された格子定数と過去に報告されていた値との間
には大きな隔たりが認められた.この原因としては,RE-J-phase のX線回折パターンが複雑でピーク
の本数や重なりが多く,各ピークを分離して回折角度を正確に測定した上で指数付けすることが非常
に難しいためであると考えられた.
本稿では,La-J-phase,および,Lu-J-phase について両者の結晶構造の違いを詳述し,RE-J-phase の
X線回折データから格子定数を算出する時の問題点について考察する.
* 東北大学多元物質科学研究所
** 東北大学学際科学国際高等研究センター
平成 15 年 12 月
希土類−シリコン−酸窒化物 RE
Si2O7N2 の結晶構造
4
2. RE‑J‑phase の結晶構造
Figs. 1 および 2 に La-J-phase(La4Si2O7N2)と Lu-J-phase(Lu4Si2O7N2)の結晶構造をそれぞれ示す.
RE-J-phase の結晶構造は Si(O,N)4 四面体が2つ結合した Si2(O,N)7 二量体と RE(O,N)(
x x = 6-8)多面体で
構成されている.La-J-phase と Lu-J-phase はいずれも空間群 P21/c(No. A-14-1)に属し,原子はすべ
て一般等価位置(4e サイト)を占め,単位格子の a-c 面内における原子配列は両者とも同じである(Figs.
1a および 2a)
.しかし,b 軸方向の積み重なり方は異なり,La-J-phase では a×b/2×c ユニットが b 軸
に沿って a/2 ずつ a 軸方向にずれているのに対し,Lu-J-phase ではそのユニットが b 軸方向に平行に並
んでいる(Figs. 1b および 2b).また,Si2(O,N)7 二量体中,2つの Si 原子を繋ぐ bridging site(O/N1)
の酸素/窒素原子占有率(Figs. 1a および 2a 中,白部/黒部の面積分率が酸素/窒素の原子占有率に
対応)は両者で異なり,La-J-phase 中の bridging site は 10%の酸素原子と 90%の窒素原子で確率的に占
有されている.これに対し,Lu-J-phase 中の bridging site は 100%窒素原子で占有されている.その結
果,La-J-phase の bridging site は5配位(2Si + 3La),Lu-J-phase では4配位(2Si + 2Lu)となっている
20)
.
RE-J-phase の結晶構造は,J-phase が生成しない RE = Eu を挟み,La4Si2O7N2 型構造を有する RE = La
– Sm のグループと Lu4Si2O7N2 型構造の RE = Gd – Lu および Y のグループに分類される 23).Figs. 3 お
よび 4 に RE-J-phase の格子定数(図中●,P21/c cell)と Shannon の希土類イオン半径 24)(+3 価;6 配
Fig. 1. Projective views of La4Si2O7N2 on the
Fig. 2. Projective views of Lu4Si2O7N2 on the
(a) a-c plane (y = -0.06 to +0.35) and (b) a-b
(a) a-c plane (y = -0.03 to +0.27) and (b) a-b plane
plane (z = -0.1 to +0.6).
20)
(z = -0.1 to +0.6).21)
高橋純一,山根久典,島田昌彦
第 59 巻 第 1,2 号
位)との関係を示す.RE = La – Sm と RE = Y, Gd – Lu のそれぞれのグループ内では,格子定数(a, b, c
軸長,およびβ角)
,単位格子体積とも希土類イオン半径の増加に伴い増加する傾向が認められる.し
かし,2つのグループ間,すなわち RE = Sm と RE = Gd の間には格子定数の変化にギャップが認めら
れる.このようなギャップを伴う格子定数の変化は,Y4Al2O9 の高温相転移に伴う格子定数の変化
と極めて類似している.Y4Al2O9 は 1370℃付
近で低温型構造(空間群 P21/c : Lu4Si2O7N2
と同型構造)− 高温型構造(空間群 P21/c:
La4Si2O7N2 と同型構造)間の可逆的なマルテ
ンサイト型相変態を生じる
25)-27)
.このとき,
格子定数は大きく変化(単位格子体積では約
0.5%)する 25),27).このような格子定数の変化
は一種の体積効果(RE-J-phase では希土類イ
オンのサイズ,Y4Al2O9 では温度による格子
の膨張)と理解されるが, RE-J-phase では
Y4Al2O9 の相変態温度に対応する RE = Eu が
存在しないことは興味深い.Y4Al2O9 高温相
と同型構造の La-J-phase は 4K においても相
変態は生じず,室温と同じ結晶構造を保って
いる 28).RE-J-phase の相転移の有無を確かめ
るためには,高温・ 低温における中性子回折
測定が必要であろう.
Fig. 3
3. RE‑J‑phase の格子定数
Figs. 3 および 4 には,過去に報告された
Fig. 4
Refined unit-cell parameters (a- and b- axes
lengths) and unit-cell volumes of RE4Si2O7N2 as a
function of ionic radii of 6-fold coordinated RE3+.
Refined unit-cell parameters (left: c-axis lengths and right: beta angle) of RE4Si2O7N2 as a function of
ionic radii of 6-fold coordinated RE3+.
marks, respectively.
The values with P21/c cell and P21/n cell are plotted with "O" and "×"
25)
平成 15 年 12 月
希土類−シリコン−酸窒化物 RE
RE-J-phase の格子定数値
20)
4),6)-19)
Si2O7N2 の結晶構造
4
を併記した.筆者らが中性子回折データの Rietveld 解析により得た
21)
La-J-phase と Lu-J-phase の格子定数と,La-J-phase と Lu-J-phase を初期構造モデルとして X 線粉末
回折データの Rietveld 解析により得た RE-J-phase の格子定数 23),および,cuspidine(Ca4Si2O7F2)を構
造モデルとして Rietveld 解析により得られた Y-J-phase の格子定数 17)を除くと,報告された格子定数値
はすべて,RE-J-phase の X 線回折パターンに cuspidine から類推された指数を付けて求められたもので
ある.筆者ら(La-J-phase, Lu-J-phase),および,MacKenzie ら(Y-J-phase)の格子定数値とその他の
報告値とは,最も初期に報告された Wills ら 6),7)の値を除くと,a, b 軸長,および,単位格子体積 V は
おおむね一致し,c 軸長,および,β角はほとんど一致していない.このような格子定数の不一致の
原因として(1)La4Si2O7N2 型構造と Lu4Si2O7N2 型構造を区別していないこと,(2)複雑な X 線回
折パターンの手作業による指数付け,
(3)誤った単位格子の取り方が挙げられる.
(1)に関しては,当時は Y4Al2O9 高温型の結晶構造が知られていなかったことや,希土類を含む
RE-J-phase の軽元素位置を粉末 X 線回折データから求めることが困難である事情からやむを得ない.
ただし,筆者らが Lu-J-phase と Lu-J-phase の構造モデルを入れ替えて Rietveld 解析を行ったところ,
得られた格子定数値は正しいモデルを用いた場合と 3σ(σは誤差)の範囲で一致していた事実から,
(1)の影響は元々小さいともいえる.
RE-J-phase は単斜晶系に属し,非常に複雑な結晶構造を呈しているため(Figs. 1 および 2)
,例えば
Fig. 5 に示す La-J-phase と Tm-J-phase の粉末 X 線回折パターンと Rietveld 解析結果 23)を見てもわかる
ように,反射の数は非常に多く,高角側ではほとんどのピークは重なっている.
(2)に関し,このよ
うな回折図形から手作業で正確にピークの回折角度(すなわち面間隔)を求め,指数を割り振ること
Fig. 5
X-ray powder diffraction patterns (CuKα) and Rietveld refinement profiles for (a) La4Si2O7N2 and (b)
Tm4Si2O7N2.
Observed (+), calculated (solid line), peak position (|), and differences. 23)
高橋純一,山根久典,島田昌彦
第 59 巻 第 1,2 号
は困難であろう.例えば Lu-J-phase の場合,2θ= 10 ゚− 50 ゚(CuKα;d = 0.8837 – 0.1823 nm)の間に
126 本のピークが現れる 21)が,Montorsi と Appendino9)の報告した X 線回折強度表には同じ 2θの範囲
に 27 本のピークしか示されていない.
Marchand ら 8)(Figs. 3 および 4 中,△印)と Montorsi ら 9),10)(同,▽印)は相補的に一連の RE-J-phase
について格子定数を報告した.筆者らと彼らが得た格子定数の希土類イオン半径依存性は,β角を除
き,おおむねその傾向は似ている.しかし,β角については,筆者らのデータは右上がりであるのに
対し,彼らのデータは右下がりのように見える.このような違いを生み出す原因が以下で示す(3)
であると考えられる.
RE-J-phase は La4Si2O7N2 型も Lu4Si2O7N2 型も空間群 P21/c(No. 14)に属する.単斜晶系の場合,主
軸(unique axis)の取り方が2種類(b 軸と c 軸)あるが,P21/c の場合はそれらに加え, 単位格子の
取り方がそれぞれ3種類ずつ(前者では P21/c,P21/n,P21/a,後者では P21/a,P21/n,P21/b)可能で
ある 29).Fig. 6 に例として P21/c 格子と P21/n 格子の関係を示す.P21/c 格子と P21/n 格子では,a 軸と
b 軸(紙面に垂直な方向)は共通であるが,c 軸とβ角は異なる値になる.また,当然ながら単位格子
体積は P21/c 格子と P21/n 格子とで等しい.P21/c 格子と P21/n 格子の格子定数は以下の関係にある.
cn = (ac2 + cc2 + 2accccosβc)1/2
(1)
βn = cos-1{(cc2 – ac2 – cn2)/(2accn)}
(2)
ここで下付きの c と n は,
それぞれ P21/c 格子と P21/n
格子を表している.Fig. 6 中に示した Lu-J-phase の
格子定数値を比較してわかるように,P21/c 格子と
P21/n 格子の c 軸長,β角値には小さいが有意な差が
認められる.Figs. 3 および 4 中に●印で示した格子
定数は P21/c 格子に基づいている.Fig. 4 中に×印で
示した値は,上式を用いて換算した P21/n 格子の格
子定数である.この図から RE = Y, Sm – Lu について
Marchand ら
8)
と Montorsi ら
9),10)
が報告した
RE-J-phase の格子定数値は,P21/c 格子ではなく P21/n
格子を単位格子としたときの値と非常に近い.P21/c
格子(Fig. 4 中,●印)と P21/n 格子(同,×印)の
差は希土類イオン半径が大きくなるほど小さくなり,
軽希土類,特に RE = Ce, Pr, Nd では過去の報告値と
Fig. 6
筆者らの結果とも近い値を示している.手作業によ
cell and P21/n cell. Values are from Lu4Si2O7N2.
Crystallographic relationship between P21/c
る格子定数計算の不正確さは,P21/n 格子と P21/c 格
子が混同され指数付けが正確に行われなかったことにより増大されたといえる.
4. おわりに
結晶構造を知ることは,その物質の基本的性質を明らかにするための第一段階である.20 世紀の材
料開発は物質創製と特性発現に主眼が置かれ,様々な物質群が見出されてきた.21 世紀に入り,材料
開発の新たな展開が期待されるが,新物質の発見や特異的な性質を発現させることはそうそう容易な
ことではない.本稿で紹介した RE-J-phase(RE4Si2O7N2, RE = Y および希土類元素)は,約 40 年の歴
史を持つ Si3N4 焼結体に関する研究の中で見出された一つの希土類酸窒化物に過ぎないが,測定技術
(X 線回折→中性子回折)や解析技術(手作業による指数付け→Rietveld 解析)の進歩により,従来
考えられていたものとは異なる結晶構造であることが今世紀になって明らかとなった一つの例である.
平成 15 年 12 月
希土類−シリコン−酸窒化物 RE
Si2O7N2 の結晶構造
4
その結果,従来は見えていなかった様々な問題点,例えば,相転移の有無,Si3N4 焼結体の高温機械強
酸窒化物における酸素/窒素の規則・ 不規則配列や Si-O/N
度に及ぼす RE-J-phase の影響とメカニズム,
結合といった結晶化学的な解釈などが浮き彫りにとなった.これはすなわち,既存・ 既知と思われる
多数の物質群の中にも様々な不確定さが隠れていることを示唆している.その意味においては,21 世
紀に行うべき重要なことの一つは 20 世紀の財産の 検証 であり,古くて新しい もの が多数存在
し,それを見出し検証することが次の問題提起となり,しいてはそれが材料開発の新たな展開に繋が
れば幸いである.
文献
(1)
(2)
三友
護: セラミックス, 38(2003), 668.
(3)
Guo, S.; Hirosaki, N.; Yamamoto, Y.; Nishimura, T.; Mitomo, M.: Scrip. Mater., 45(2001), 867.
(4)
Nishimura, T.; Mitomo, M.: J. Mater. Res., 10(1995), 240.
(5)
Hirosaki, N.; Yamamoto, Y.; Nishimura, T.; Mitomo, M.; Takahashi, J.; Yamane, H.; Shimada, M.: J.
Nishimura, T.; Mitomo, M.; Suematsu, H.: J. Mater. Res., 12(1997), 203.
Am. Ceram. Soc., 85(2002), 2861.
(6)
(7)
Wills, R. R.; Stewart, R. W.; Cunningham J. A.; Wimmer, J. M.: J. Mater. Sci., 11(1976), 749.
Wills, R. R.; Holmquist, S.; Wimmer, J. M.; Cunningham, J. A.: J. Mater. Sci., 11(1976), 1305.
(8)
Marchand, M. M. R.; Jayaweera, A.; Verdier, P.; Lang, J.: Compt. Rend. Acad. Sci. Paris, Ser. C,
283(1976), 675.
(9)
Montorsi, M.; Appendino, P.: J. Less-Comm. Metals, 68(1979), 193.
(10)
Montorsi, M.; Appendino, P.: Am. Ceram. Soc. Bull., 58(1979), 789.
(11)
(12)
Morgan, P. E. D.: J. Am. Ceram. Soc. 62(1979), 636.
Ii, N.; Mitomo, M.; Inoue, Z.: J. Mater. Sci. 15(1980), 1691.
(13)
Mitomo, M.; Izumi, F.; Horiuchi, S.; Matsui, Y.: J. Mater. Sci. 17(1982), 2359.
(14)
Guha, J. P.: J. Mater. Sci. 15(1980), 262.
(15)
Rae, A. W. J. M.; Buang, K. B.; Thompson, D. P.: J. Mater. Sci. 15(1980), 264.
(16)
Thompson, D. P.: Tailoring Multiphase and Composite Ceramics, Materials Science Research,
(17)
20(1986), Plenum, NY.
MacKenzie, K. J. D.; Gaonsfold, G. J.; Tyan, M. J.: J. Eur. Ceram. Soc. 16(1996), 553.
(18)
Liddell, K.; Thompson, D. P.; Wang, P. L.; Sum, W. Y.; Gao, L.; Yan, D. S.: J. Eur. Ceram. Soc.
18(1998), 1479.
(19)
Ijevskii, V. A.; Kolitsch, U.; Seifert, H. J.; Wiedmann, I.; Aldinger, F.: J. Eur. Ceram. Soc. 18(1998),
543.
(20)
Takahashi, J.; Yamane, H.; Hirosaki, N.; Yamamoto, Y.; Suehiro, T.; Kamiyama, T.; Shimada, M.:
Chem. Mater. 15(2003), 1099.
(21)
Takahashi, J.; Yamane, H.; Shimada, M.; Yamamoto, Y.; Hirosaki, N.; Mitomo, M.; Oikawa, K.; Torii,
S.; Kamiyama, T.: J. Am. Ceram. Soc. 85(2002), 2072.
(22)
Takahashi, J.; Yamane, H.; Yamamoto, Y.; Hirosaki, N.; Mitomo, M.; Oikawa, K.; Torii, S.;
(23)
Takahashi, J.; Yamane, H.; Hirosaki, N.; Yamamoto, Y.; Mitomo, M.; Shimada, M.: J. Euro. Ceram.
Soc. (2004) in press.
(24)
Shannon, R. D.: Acta Cryst. A32(1976), 751.
(25)
Yamane, H.; Shimada, M.; Hunter B. A.: J. Solid State Chem. 141(1998), 466.
(26)
Yamane, H.; Omori, M.; Okubo, A.; Hirai, T.: J. Am. Ceram. Soc. 76(1993), 2382.
Kamiyama, T.; Shimada, M.: Key Engineering Materials 237-237(2003), 53.
高橋純一,山根久典,島田昌彦
第 59 巻 第 1,2 号
(27)
Yamane, H.; Ogasawara, K.; Omori, M.; Hirai, T.: J. Am. Ceram. Soc. 78(1995), 1230.
(28)
Takahashi, J., unpublished work
(29)
International Tables for Crystallography, Vol. A, Fourth Revised Edition, The Netherlands, 1996.
平成 16 年 2 月 20 日 印刷
平成 16 年 3 月
3 日 発行
東北大学多元物質科学研究所
素材工学研究棟
発 行 者
所
長
中 西 八 郎
〒980-8577 仙台市青葉区片平二丁目 1 番 1 号
電話 022 (217) 5204 (総務課庶務掛)
FAX 022 (217) 5211
印 刷 所
株式会社
セ
ン
ト
〒980-0011 仙台市青葉区上杉一丁目 17 番 20 号
電話 022 (261) 6688 (代表番号)
印 刷 者
永
野
行
雄
<非 売 品 >
本彙報に掲載の論文報告等は発行者の承諾なくして他に転載することを禁ずる
© Copyright 2026 Paperzz