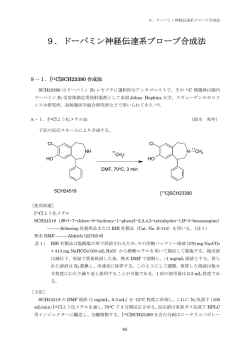
13.その他の神経伝達系プローブ合成 13.その他の神経伝達系プローブ合成法 13-1.[11C]ドキセピン合成法 ドキセピンは三環系抗うつ薬であり、様々な病気の治療薬として用いられている。11C−標識 された[11C]ドキセピンはヒスタミン作動性神経細胞におけるシナプス後膜にあるヒスタミン H1 受容体に結合し、脳内のヒスタミン H1 受容体測定の放射性薬剤として利用される 1)。 A-1.[11C]メチルトリフレートによるループ合成法 (加藤 元久) 下記の反応スキームにより合成する。 11 H N CH3 CH3 N 11CH OTf 3 CH3 NaOH/2-butanone O O [使用試薬] [11C]メチルトリフレート NordoxepinHCl ———Sigma(N-0392) 1.2 M NaOH ———試薬特級 2−Butanone(MEK)——— Aldrich(270695) 局方 25%アスコルビン酸注射液———扶桑薬品 [方法] NordoxepinHCl(約 1 mg)を MEK(60 L)に溶かし、これに 1.2 M NaOH(2 L)を加 える。pH 試験紙を用いてこの反応溶液がアルカリ(pH 9 以上)であることを確認する。反応 溶液を反応ループに注入し、He 気流下(50 mL/min)[11C]メチルトリフレートを通し、これ に捕集する(註 1)。反応溶液を固相抽出カラムで濃縮し(註2)、HPLC インジェクターに導 入の後、分取カラムで分離精製する。その分取液に蒸留水(30 mL)を加え、Sep-Pak Plus tC18 に通して目的物を捕集分離する(註3)。エタノールで捕集物を溶出し、ロータリエバポレータ ーでエタノールを留去する(註4) 。生理食塩水を加えて残渣を溶かし、メンブレンフィルター を通して無菌バイアルに捕集する。 註1) 1-6オンカラム標識法とループ標識法のループ標識法を参照。 註2) 1-7分取 HPLC カラムへの反応液自動注入法の固相抽出カラム濃縮法を参照。 151 註3) 1-8HPLC 分離精製物の固相抽出調製法を参照。 註4) 放射線分解を抑制するため、留去前に 0.2 mL の注射用アスコルビン酸を加えておく。 [HPLC 分取条件] カラム:YMC-Pack ODS-A(内径 10 mm X 長さ 250 mm)、ワイエムシー 溶離液:CH3CN/50 mM AcONH4(43/57) 速:5 mL/min 流 検出器:UV(254 nm)、放射能検出器 溶出時間:前駆体 4.5 分;目的物 9.5 分 Radioactivity 10 9 8 7 6 5 4 3 Nordoxepin 2 UV Doxepin 1 0 0 3 6 9 Elution time (min) 12 15 A-2.[11C]メチルトリフレートによる液相法 (石渡 喜一) 下記の反応スキームにより合成する。 11 H N N 11CH OTf 3 CH3 CH3 CH3 NaOH/acetone O O [使用試薬] [11C]メチルトリフレート NordoxepinHCl ———Sigma (N-0392) アセトン———試薬特級 水酸化ナトリウム———試薬特級 局方 25%アスコルビン酸注射液 [方法] 1 M NaOH(10 L)を含む nordoxepinHCl のアセトン溶液(1 mg/mL、0.25 mL)(註1) 152 13.その他の神経伝達系プローブ合成 に、室温下で He 気流下(30-50 mL/min)[11C]メチルトリフレートを通して捕集する。直ちに 反応液に 1.3 mL の HPLC 溶璃液を加えて希釈し、HPLC により分離精製する。 以下の処理及び注意点は、A−1に準ずる。 註1) NordoxepinHCl のアセトン溶液は、数ヶ月は室温で保存して使用することができる。 B.分析法 [放射化学的純度] HPLC カラム:TSK-GEL ODS-80TM(内径 8 mm X 長さ 100 mm) 、東ソー 溶離液:CH3CN/180 mM HCO2NH4(45/55) 流 速:1.5 mL/min 検出器:UV(254 nm)、放射能検出器 保持時間:7.0 分 C.その他 [急性毒性]2) LD50 マウス:96 mol/26 mg/kg (i.v.) ラット:59 mol 16 mg)/kg (i.v.) (Merck Index 11th Ed.) [被曝線量]3) 臓器 線量(Gy/MBq) 臓器 線量(Gy/MBq) 脳 18 腎臓 42 肝臓 10 腸 3.3 脾臓 51 赤髄 1.3 睾丸 0.70 膀胱壁 16 胃壁 3.1 肺臓 31 全身の線量当量は 6.9 Sv/MBq 参考文献 1. Yanai K., Watanabe T., Yokoyama H., et al.: Neurosci. Lett., 137, 145−148 (1992). 2. 東北大学サイクロトロン製造放射性薬剤品質管理基準 3. Nakamura T., Hayashi Y., Watabe H., et al.: Phys. Med. Biol., 43, 389−405 (1998). 13-2.[11C]SA4503 合成法 (石渡 喜一) [11C]SA4503(1−(3,4−dimethoxyphenethyl)−4−(3−phenylpropyl)piperazine)はシグマ 1 受 153 容体に高い親和性を示すリガンドである 1-4)。シグマ 1 受容体は脳内に広く分布し、アルツハ イマー病、認知症、鬱病、統合失調症、不安神経症等の中枢神経疾患と関連した受容体であ ることが明らかとなり、新しい PET 診断法として期待され 5)、2000 年 6 月より臨床研究が 開始された 6-8)。また、ハロペリドールなど多くの向精神薬はシグマ受容体への親和性を有し、 これら薬剤の受容体占拠率と薬理効果の関係を PET により明らかにすることが可能になる 9-11)。 A−1.[11C]よう化メチル法 下記の反応スキームにより合成する 1)。 N 11 N OCH3 CH3 I NaOH, DMF OH N N OCH3 O11 CH3 [使用試薬] [11C]よう化メチル デ メ チ ル SA4503 ( 1−(4−hydroxy−3−methoxyphenethyl)−4−(3−phenylpropyl) piperazine 2HCl)(註1) 無水 DMFAldrich(22705-6) NaHAldrich 註1) 合成は Fujimura らの方法による 12)。 [方法] NaH(1-2 mg)を含むデメチル SA4503 の DMF 溶液(1 mg/mL、0.25 mL)を–15ºC に冷 却し、これに He 気流下(30-50 mL/min)[11C]よう化メチルを通して捕集する。120ºC で 1 分 間反応させた後、反応液に H2O で 2 倍希釈した HPLC 溶離液(1.3 mL)を加えて希釈し、HPLC により分離精製する。 分離された[11C]SA4504 溶液は、ロータリエバポレーターのフラスコに分取し、溶媒を除い た後、注射用生理食塩水に溶解する。 (註1)メンブレンフィルターを通して無菌バイアルに捕 集する。(註2) 註1) [11C]SA4503 溶液は濃縮、乾固により多少の分解が認められるが、前もってアスコルビ ン酸(0.2 mL の 100 mg/mL の注射用アスコルビン酸水溶液)を加えておくことで分 解を抑えることができる。 註2) メンブレンフィルターには、脂溶性薬剤の吸着の少ないマイレックス GV フィルターTM (ミリポア)などを使用する。 A−2.[11C]メチルトリフレート法 次の反応スキームにより合成する 13)。 154 13.その他の神経伝達系プローブ合成 N 11 N OCH3 CH3OTf N N NaOH, DMF OH OCH3 O11 CH3 [使用試薬] [11C]メチルトリフレート デメチル SA4503 DMF特級試薬 水酸化ナトリウム特級試薬 [方法] NaOH(5 L)を含むデメチル SA4503 の DMF 溶液(1 mg/mL、0.25 mL)を–15ºC 冷却 し、これに He 気流下(30-50 mL/min)[11C]メチルトリフレートを通して捕集する。120ºC で 1分間反応させた後、反応液に H2O で 2 倍希釈した HPLC 溶離液(1.3 mL)を加えて希釈し、 HPLC により分離精製する。 以下の処理及び注意点は、[11C]よう化メチル法に準ずる。 [合成法の特徴と問題点] [11C]メチルトリフレートとの反応では、通常溶媒として汎用されるアセトンへのデメチル SA4503 の溶解度が低く、溶媒として DMF を用いた。DMF 溶媒では、室温下で[11C]メチルト リフレートを通じただけでは、[11C]SA4503 の合成収率は低く、120℃で1分間の加熱が必要で あった。 [HPLC 分取条件](註1) カラム:YMC-Pack ODS-A(内径 10 mm×長さ 250 mm)、ワイエムシー 溶離液:CH3CN/50 mM AcOH/50 mM AcONH4(35/32.5/32.5) 流 速:5 mL/min 検出器:UV(280 nm)、線検出器 保持時間:原料 5.0 分、目的物 7.5 分 155 B.分析法 [放射化学的純度] HPLC i) カラム:TSKgel Super-ODS(内径 4.6 mm×長さ 100 mm) 、東ソー 溶離液:CH3CN/50 mM AcOH/50 mM AcONH4(30/35/35) 流 速:1 mL/min 検出器:UV、280 nm、線検出器 保持時間:目的物 4.6 分、原料 4.2 分 ii) カラム:TSKgel ODS-140HTP(2.1 mm×長さ 50 mm、2.3 m)、東ソー 溶離液:CH3CN/50 mM AcOH/50 mM AcONH4(25/37.5/37.5) 流 速:0.5 mL/min 検出器:UV、280 nm、線検出器 保持時間:原料 1.5 分、目的物 2.0 分 C.その他 [被曝線量]14) ヒト全身 PET 動態計測による。 実効線量:6.9 Sv/MBq 甲状腺:24.1 Gy/MBq、脾臓:23.6 Gy/MBq、肺:22.1 Gy/MBq 全身:2.9 Gy/MBq [急性毒性]14) LD50:マウス雄、437 mol (193 mg)/kg (p.o.) 雌、356 mol (157 mg)/kg (p.o.)(未発表データ) [11C]SA4503 の担体量効果を調べる実験において、SA4503 を 4 匹の雄マウスに 5 mol (2.2 mg)/kg を静脈注射したところいずれのマウスも死亡しなかった。また、[11C]SA4503 の PET 実験において、非放射体をネコに 2.3 mol (1.0 mg)/kg、サルに 1.9 mol (0.86 mg)/kg を負荷 したとき、特に異常な所見は観察されなかった。 [薬理作用]14) SA4503 は坑健忘効果、坑うつ効果、神経保護効果を有する。 体温低下:100 mg/kg、振せん誘発:40 mg/kg、瞳孔縮小誘発:50 mg/kg など。 参考文献 1. Kawamura K., Ishiwata K., Tajima H., et al.: Nucl. Med. Biol., 27, 255–261 (2000). 2. Kawamura K., Ishiwata K., Shimada Y., et al.: Ann. Nucl. Med., 14, 285–292 (2000). 3. Ishiwata K., Tsukada H., Kawamura K., et al.: Synapse, 40, 235–237 (2001). 4. Kawamura K., Kimura Y., Tsukada H., et al.: Neurobiol. Aging, 24, 745–752 (2003). 5. Hashimoto K., Ishiwata K.: Curr. Pharm. Design, 12, 3857–3876 (2006). 156 13.その他の神経伝達系プローブ合成 6. Mishina M., Ishiwata K., Ishii K., et al.: Acta Neulologica, 112, 103–107 (2005). 7. Sakata M., Kimura Y., Naganawa M., et al.: NeuroImage, 35, 1–8 (2007). 8. Mishina M., Ohyama M., Ishii K., et al.: Ann. Nucl. Med., 22: 151–156 (2008). 9. Ishiwata K., Oda K., Sakata M., et al.: Ann. Nucl. Med., 20, 569–573 (2006) 10. Ishikawa M., Ishiwata K., Ishii K., et al.: Biol. Psychiatry, 62, 878–883 (2007). 11. Ishikawa M., Sakata M., Ishii K., et al.: Int. J. Neuropsychopharm., 12, 1127–1131 (2009). 12. Fujimura K., Matsumoto J., Niwa M., et al.: Bioorg. Med. Chem., 5, 1675–1683 (1997). 13. Kawamura K., Ishiwata K.: Ann. Nucl. Med., 18, 165–168 (2004). 14. 東京都健康長寿医療センター研究所附属診療所短寿命放射性薬剤臨床利用委員会資料 13-3.[11C]MPDX 合成法 (石渡 喜一) [11C]MPDX([3−methyl−11C]8−dicyclopropylmethyl−3−methyl−1−propylxanthine)はキサ ンチン構造を持ち、アデノシン A1 受容体に高い親和性を示すリガンドである 1,2)。アデノシン A1 受容体は脳に広く発現しており、アルツハイマー病やてんかん等の神経変性疾患や脳虚血を 診断する薬剤として期待され 3)、2002 年 6 月より臨床研究が開始された 4,5,6)。 A.合成法 下記の反応スキームにより合成する。 O O H N HN O N N 11 CH3I NaH, DMF H311C O H N N N N C3H7 C 3H7 [使用試薬] [11C]よう化メチル デメチル MPDX(註1) 無水 DMFAldrich(22705-6) NaHAldrich 註1) 8−dicyclopropylmethyl−1−propylxanthine。合成は Shimada らの方法による 6)。 [方法] NaH(1~2 mg)を含むデメチル MPDX の DMF 溶液(1 mg/mL、0.25 mL)を–15C に冷 却し、これに He 気流下(30~50 mL/min)[11C]よう化メチルを通して捕集する。120C で 1 分間 反応させた後、反応液に 0.1 M HCl で 2 倍希釈した HPLC 溶離液(1.3 mL)を加えて希釈し、 157 HPLC により分離精製する。(註1) 註1) メンブレンフィルターには、脂溶性薬剤の吸着の少ないマイレックス GV フィルターTM (ミリポア)などを使用する。 [合成法の特徴と問題点] 本方法では、目的の[11C]MPDX の他に少量の 7−[11C]メチル体が合成される。この 7−異性体 のアデノシン A1 受容体に対する親和性は[11C]MPDX より低いが、両者を確実に分離するため、 比較的大きめのカラムを用いている。また、DMF の無水状態の十分でないと 7−異性体の割合 は増加する。少量の NaOH を含む DMF 溶液中での[11C]メチルトリフレートとの反応でも、 [11C]MPDX は 34%の放射化学的収率で得られたが、7−[11C]メチル体の割合も 15%に達した 7)。 [HPLC 分取条件] カラム:YMC-Pack ODS-A(内径 20 mm X 長さ 150 mm)、ワイエムシー 溶離液:CH3CN/H2O(45/55) 流 速:15 mL/min 検出器:UV(260 nm)、線検出器 保持時間:原料 5.1 分、目的物 8.0 分 B.分析法 [放射化学的純度] HPLC i) カラム:TSKgel Super-ODS(内径 4.6 mm X 長さ 100 mm) 、東ソー 溶離液:CH3CN/H2O(40/60) 流 速:1 mL/min 検出器:UV(260 nm)、線検出器 保持時間:原料 2.4 分、目的物 3.8 分 ii) カラム:TSKgel ODS-140HTP(内径 2.1 mm×長さ 50 mm、2.3 m)、東ソー 溶離液:CH3CN/H2O(30/70) 流 速:0.5 mL/min 検出器:UV(260 nm)、線検出器 保持時間:原料 1.1 分、目的物 2.4 分 158 13.その他の神経伝達系プローブ合成 C.その他 [被曝線量]9) ヒト全身 PET 動態計測による。 実効線量:7.7 Sv/MBq 小腸:22.1 Gy/MBq、肝臓:11.2 Gy/MBq、心臓壁:8.9 Gy/MBq 全身:2.9 Gy/MBq [急性毒性]2) 雌雄それぞれ 5 匹のラットに、臨床想定の最大投与量の 740 MBq/74 nmol/60kg(最低の比 放射能 10 TBq/mmol として)の 10,000 倍にあたる 12.3 mol/3.73 mg/kg の MPDX を単回投 与(腹腔)して、30 分、1、3、6 時間後、その後 14 日まで 1 日 1 回観察したが、死亡するも のはなく、一般状態に異常を認めなかった。体重増加も正常であり、第 15 日に病理学的検査を おこなった結果、いずれの動物にも異常所見は認められなかった。LD50:ラット(雌雄、腹腔)、 >12.3 mol/3.73 mg/kg [11C]MPDX の臨床用調製薬剤 3 ロットについて、雌雄各 5 匹のラットに臨床想定の最大投与 量(740 MBq)の 40 倍量(500 MBq/kg)を静脈投与し、上記と同様に 14 日間の観察と病理 学的検査をおこなったとき、いずれの動物にも異常所見は認められなかった。 [突然変異試験(Ames 試験)]2) S. Typhimurium TA98、TA100、TA1535 および TA1537 を用いて調べた MPDX の復帰突 然変異原性は、5,000 g/plate 以下のアッセイで陰性であった。 参考文献 1. Noguchi J., Ishiwata K., Furuta R., et al.: Nucl. Med. Biol., 24, 53–59 (1997). 2. Ishiwata K., Nariai T., Kimura Y., et al.: Ann. Nucl. Med., 16, 377–382 (2002). 3. Nariai T., Shimada Y., Ishiwata K., et al. J. Nucl. Med., 44, 1839–1844 (2003). 4. Fukumitsu N., Ishii K., Kimura Y., et al.: Ann. Nucl. Med., 17, 511–515 (2003). 5. Fukumitsu N., Ishii K., Kimura Y., et al.: J. Nucl. Med., 46, 32–37 (2005). 6. Fukumitsu N., Ishii K., Kimura Y., et al.: Ann. Nucl. Med., 22, 841–847 (2008). 7. Shimada J., Suzuki J., Nonaka H., et al.: J. Med. Chem., 35, 924–930 (1992). 8. Kawamura K., Ishiwata K.: Ann. Nucl. Med., 18, 165–168 (2004). 9. 東京都老人総合研究所附属診療所短寿命放射性薬剤臨床利用委員会資料 13-4.[11C]TMSX 合成法 (石渡 喜一) [11C]TMSX([7−methyl−11C](E)−8−(3,4,5−trimethoxystyryl)−1,3,7−trimethylxanthine、別 名[11C]KF18446)はキサンチン構造を持ち、アデノシン A2A 受容体に高い親和性を示すリガン ドである 1-3 )。アデノシン A2A 受容体は脳ではドーパミン D2 受容体と同じ GABAergic– 159 enkephaline ニューロンに発現しており、パーキンソン症候群などのポストシナプスの変性や 脱落あるいは統合失調症等の診断する薬剤として期待される。2003 年 2 月より臨床研究が開始 され 4-6)、心筋や骨格筋のアデノシン A2A 受容体イメージングや機能評価へも応用可能である 7,8)。 [11C]TMSX は水溶液中で光により異性化するので、合成から臨床使用に至る全ての行程で注意 を要する。 A−1.[11C]よう化メチル法 1,3) 下記の反応スキームにより合成する。 O H3C O N N O H N N CH3 OCH3 OCH3 11 H3C CH3I Cs 2CO3, DMF O OCH3 11 CH3 N N N N CH3 OCH3 OCH3 OCH3 [使用試薬] [11C]よう化メチル デメチル TMSX(註1) 無水 DMFAldrich(22705-6) 炭酸セシウム ポリソルベ—ト 80(polyoxyethylene(20) sorbitan monooleate)和光純薬工業製 註1) 合成は Shimada らの方法による 7)。最近、ParmaSynth 社が販売している。 [方法](註1) 炭酸セシウム(5~10 mg)を入れた遮光した反応容器にデメチル TMSX の DMF 溶液(1 mg/mL、0.25 mL)を入れ、120C で 1 分間加熱する。この反応溶液を–15C に冷却し、これ に He 気流下(30~50 mL/min)[11C]よう化メチルを通して捕集する。120C で 3 分間反応さ せた後、反応液に 0.1 M HCl で 2 倍に希釈した HPLC 溶離液(1.3 mL)を加えて希釈し、HPLC により分離精製する。 分離された[11C]TMSX 溶液は、ロータリエバポレーターのフラスコに分取し、溶媒を除いた 後、注射用生理食塩水に溶解する。 (註2、3)メンブレンフィルターを通して無菌バイアルに 捕集する。(註4) 註1) TMSX およびデメチル TMSX のスチリル基は、水溶液では光により異性化するので、 すべての操作はなるべく光を避けて行うことが望ましい。 註2) [11C]TMSX 溶液は濃縮、乾固により多少の分解が認められるが、前もってアスコルビ ン酸(0.2 mL の 100 mg/mL の注射用アスコルビン酸水溶液)を加えておくことで分 解を抑えることができる。 註3) 生理食塩水は 0.25%ポリソルベート 80 を含む。 註4) メンブレンフィルターには、脂溶性薬剤の吸着の少ないマイレックス GV フィルターTM (ミリポア)などを使用する。 160 13.その他の神経伝達系プローブ合成 A−2.[11C]メチルトリフレート法 10) 下記の反応スキームにより合成する。 O H3C N N N N CH3 O O H OCH3 OCH 3 11 H3C CH3OTf Cs 2CO3, DMF N O OCH3 11 CH3 N N N CH3 OCH3 OCH3 OCH3 [使用試薬] [11C]メチルトリフレート デメチル TMSX 無水 DMFAldrich(22705-6) 炭酸セシウム [方法](註1) 炭酸セシウム(5~10 mg)を入れた遮光した反応容器にデメチル TMSX の DMF 溶液(1 mg/mL、0.25 mL)を入れ、120C で1分間加熱する。この反応溶液を–15C に冷却し、He 気流下(30~50 mL/min)[11C]メチルトリフレートを通して捕集する。120C で 1 分間反応さ せた後、反応液に 0.1 M HCl で 2 倍に希釈した HPLC 溶璃液(1.3 mL)を加えて希釈し、HPLC により分離精製する。 以下の処理及び注意点は、[11C]よう化メチル法に準ずる。 [合成法の特徴と問題点] E−体(trans 体)である[11C]TMSX は水溶液中で光により異性化し、E−体と R−体(cis 体) の平衡状態になる。R−体はアデノシン A2A 受容体に対する親和性は E−体より低いので、HPLC 分離以後は、光をなるべく遮断する必要がある。 [11C]メチルトリフレートとの反応では、通常溶媒として汎用されるアセトンへのデメチル TMSX の溶解度が低く、溶媒として DMF を用いた。また、アルカリとして NaOH を用いたと きは、[11C]TMSX の放射化学的収率は炭酸セシウムを用いたときより低下した 10)。 [HPLC 分取条件] カラム:YMC-Pack ODS-A(内径 10 mm X 長さ 250 mm)、ワイエムシー 溶離液:CH3CN/H2O(50/50) 流 速:5 mL/min 検出器:UV(260 nm)、線検出器 保持時間:原料 3.9 分、目的物 5.7 分 161 B.分析法 [放射化学的純度] HPLC i) カラム:TSKgel Super-ODS(内径 4.6 mm X 長さ 100 mm) 、東ソー 溶離液:CH3CN/H2O(40/60) 流 速:1 mL/min 検出器:UV(260 nm)、線検出器 保持時間:原料 2.4 分、目的物 5.5 分 ii) カラム:TSKgel ODS-140HTP(内径 2.1 mm×長さ 50 mm、2.3 m)、東ソー 溶離液:CH3CN/H2O(25/75) 流 速:0.1 mL/min 検出器:UV(260 nm)、線検出器 保持時間:原料 1.3 分、目的物 2.6 分 C.その他 [被曝線量]11) ヒト全身 PET 動態計測による。 実効線量:3.6 Sv/MBq 肝臓:11.0 Gy/MBq、胆嚢壁:11.0 Gy/MBq、腎臓:9.2 Gy/MBq 全身:2.9 Gy/MBq [急性毒性]3) 雌雄それぞれ 5 匹のラットに、臨床想定の最大投与量の 740 MBq/74 nmol/60kg(最低の比 放射能 10 TBq/mmol として)の 10,000 倍にあたる 12.3 µmol/4.77 mg/kg の TMSX を単回投 与(腹腔)して、30 分、1、3、6 時間後、その後 14 日まで 1 日 1 回観察したが、死亡するも のはなく、一般状態に異常を認めなかった。体重増加も正常であり、第 15 日に病理学的検査を おこなった結果、いずれの動物にも異常所見は認められなかった。 LD50:ラット(雌雄、腹腔)、>12.3 µmol/4.77 mg/kg [11C]TMSX の臨床用調製薬剤 3 ロットについて、雌雄各 3 匹のラットに臨床想定の最大投与 量(740 MBq)の 100 倍量(1.23 GBq/kg)を静脈投与し、上記と同様に 14 日間の観察と病理 学的検査をおこなったとき、いずれの動物にも異常所見は認められなかった。 162 13.その他の神経伝達系プローブ合成 [突然変異試験(Ames 試験)]3) S. Typhimurium TA98、TA100、TA1535 および TA1537 を用いて調べた TMSX の復帰突然 変異原性は、5,000 g/plate 以下のアッセイで陰性であった。 参考文献 1. Ishiwata K., Noguchi J., Wakabayashi S., et al.: J. Nucl. Med., 41, 345–354 (2000). 2. Ishiwata K., Ogi N., Shimada J., et al.: Ann. Nucl. Med., 14, 81–89 (2000). 3. Ishiwata K., Wang W.F., Kimura Y., et al.: Ann. Nucl. Med., 17, 205–211 (2003). 4. Ishiwata K., Mishina M., Kimura Y., et al.: Synapse, 55, 133–136 (2005). 5. Mishina M., Ishiwata K., Kimura Y., et al.: Synapse, 61, 778–784 (2007). 6. Naganawa M., Kimura Y., Mishina M., et al.: Eur. J. Nucl. Med. Mol. Imaging, 34, 679–787 (2007). 7. Ishiwata K, Mizuno M, Kimura Y., et al.: Nucl. Med. Biol., 31, 949–956 (2004). 8. Mizuno M., Kimura K., Tokizawa K., et al.: Nucl. Med. Biol., 32, 831–836 (2005). 9. Shimada J., Suzuki F., Nonaka H., et al.: J. Med. Chem., 35, 2342–2345 (1992). 10. Kawamura K., Ishiwata K.: Ann. Nucl. Med., 18, 165–168 (2004). 11. 東京都老人総合研究所附属診療所短寿命放射性薬剤臨床利用委員会資料 13-5.S−(−)−[11C]CGP-12177 合成法 (西嶋 剣一、久下 裕司) S−(−)− CGP-12177 は、親水性の非選択的アンタゴニスト(部分的アゴニスト)である。こ の化合物は、1)受容体に対する親和性が高いこと、2)脂溶性が低く非特異的な結合が少ない こと、3)細胞内に取り込まれないため、膜表面受容体のリガンドとみなせることなどの優れた 特徴を有している。その 11C-標識された S−(−)−[11C]CGP-12177 は心臓や肺の受容体密度測定 に有効であると報告されている 1)。 A.合成法 下記の反応スキームにより合成する 2-7)。 t-Bu N H HO t-Bu O H 11 COCl2 NH 2 N H HO O H H N 11 C O N H NH 2 [使用試薬] [11C]ホスゲン(註1) (2S)−1−(2−Amino−3−nitrophenoxy)−3−(t−butylamino)−2−propanol(アミノニトロ体) (註2) 163 (2S)−1−(2,3−Diaminophenoxy)−3−(t−butylamino)−2−propanol(ジアミノ前駆体)(註3) トルエン———和光純薬(有機合成用:209-13445) Palladium/carbon———和光純薬(5% Pd:165-07542) 註1) 1-5[11C]ホスゲンの合成参照。 註2) セティカンパニー経由で ABX に受託合成できる。価格は、500 mg が 80 万円、1 g が 110 万円、2 g が 160 万円(各税別)であり、納期は 3 ヶ月程度となっている。 註3) アミノニトロ体を 5% palladium/carbon、H2 による還元反応により合成する 5,7)。室温 下、5 時間程度で定量的に反応は進行する。反応後、palladium/carbon をフィルター ろ過し、溶媒を除去すると無色のペースト状でジアミノ前駆体が得られる。このジア ミノ前駆体の再結晶は行わず、このまま標識反応に用いる。ジアミノ前駆体は、不安 定なため、空気や時間の経過により黒く変色するので、前駆体合成時や保存時に注意 を要する。ジアミノ前駆体が黒く着色すると収量の低下、比放射能の低下が観察され る。保存状態がよければ、6 ヶ月程度の使用は可能である。 [方法] ジアミノ前駆体(3 mg 程度)をトルエン(0.5 mL)に溶解させる(註1)。この溶液を反応 容器に加え、[11C]ホスゲンを吹き込んで反応する(註2)。反応終了後、トルエン溶媒を減圧下 で留去し(註3)、HPLC 溶離液(1.7 mL)に溶解して HPLC により分離精製する。溶出した 目的分画をロータリエバポレーターに導入し、エタノールを減圧除去したのち(註4)、注射用 生理食塩水に溶かし、最終的に 0.22 m のメンブレンフィルターを通して注射用薬剤とする(註 5)。 註1) ジアミノ前駆体は、ペースト状のため、スパーテルでかきとる。目安としてトルエン 溶媒に黄色がつく程度で十分である。溶液の色が黄暗色の場合は、収量の低下の恐れ がある。 註2) 室温下で[11C]ホスゲンを捕集している。反応容器の放射能センサー値が最大になった ところで He を止め反応を終了する。 註3) トルエンを完全に除去してしまうと、反応容器から放射性化合物が移送しにくくなる。 トルエンは、完全に除去せずある程度残し、カラムへインジェクトする。カラムはエ タノールにより洗浄することによりトルエンを除く。 註4) 蒸 発 乾 固 さ せ て し ま う と 、 生 理 食 塩 水 で は ガ ラ ス 容 器 の 壁 に 吸 着 し た S−(−)− [11C]CGP-12177 が溶解されにくくなるので注意する。したがって、製剤中には、微量 のエタノールが含まれるため濃度を測定する必要がある 7)。 註5) 本製剤の pH は HPLC 溶出溶媒の pH が 2.3 のため、4.0 と酸性側を示す。そのため「放 射性薬剤の基準と臨床使用の指針」に従い最終的に日本薬局方炭酸水素ナトリウム注 射液を加えることにより pH を 7.0 とする 7)。 [その他の注意事項] 合成前後において、不活性ガスによる十分なパージを行う。 [11C]メタンの捕集が低下したときは、Porapak Q カラムのエージングを行ことで改善する。 Cl2 を使用するため、電磁弁の故障が考えられる。そのため塩素ガスが通じるラインは、不活 164 13.その他の神経伝達系プローブ合成 性ガスで置換しておくこと。 Cl2 の採取は、ディスポーザブルタイプの 10 mL シリンジを用いている。 アンチモンカラムは、10 回程度の使用が可能である。 [合成法の特徴と問題点] S−(−)−[11C]CGP-12177 の合成は、[11C]ホスゲン合成の成否が鍵となる。収量が低下した場 合は[11C]ホスゲンの収量が低く、1)[11C]四塩化炭素が圧倒的に多い場合と、2)[11C]四塩化 炭素も少ない場合の 2 つのパターンがある。1)の場合は、[11C]ホスゲンが生成していないた め、鉄顆粒−酸化鉄粉末カラムの不具合を考え、カラムの調製や電気炉の調整が必要となる。2) の場合は、[11C]ホスゲンの分解が推定され、装置内の水分が原因と考えられる。この場合はラ インのパージを行うなど水分の除去を行うことが肝要である。 [HPLC 分取条件] カラム:Megapak SIL C18-10(内径 7.5 mm X 長さ×250 mm) 、日本分光 溶離液:EtOH/0.9% NaCl/85% H3PO4(200/800/1.7)pH 2.3(註1) 流 速:3 mL/min 検出器:UV(254 nm)、放射能検出器 溶出時間:前駆体 4.0 分、目的物 7.0 分 註1) 0.9% NaCl は注射用の生理食塩水を用いるが、注射用蒸留水でも可。 Radio UV (254 nm) ジアミノ体 [11C]CGP12177 0 10 5 165 min B.分析法 [放射化学的純度] HPLC カラム:Finepak SIL C18S(内径 150 mm X 長さ 4.6 mm) 、日本分光 溶離液:EtOH/0.9% NaCl/85% H3PO4(200/800/1.7)pH 2.3(註1) 流 速:0.5 mL/min 検出器:UV(254 nm)、放射能検出器 溶出時間:6.0 分 註1) 0.9% NaCl は注射用生理食塩水を用いるが、注射用蒸留水でも可。 [化学的純度] エタノールの分析濃度 GC カラム:TSG-1 15% Shincarbon A 60/80 glass column; (ガラスカラム、内径 3.2 mm X 長さ 3.1 m)、信和化工 検出器:FID 検出器(カラムオーブン温度 120C、カラム温度 90°C、インジェクター 温度 180°C、FID 温度 180°C;H2 50 kPa、air 50 kPa、He 50 kPa) C.その他 [毒性] 参考:LD50、ラット 104~135 mg/kg(Dr. C. Crouzel より提供された資料より) [被曝線量] 臓器 線量(Gy/MBq) 臓器 線量(Gy/MBq) 副腎 5.99 筋肉 2.85 脳 8.73 卵巣 5.21 胸部 4.92 膵臓 6.03 胆嚢 5.15 赤色髄 5.00 小腸壁 4.78 骨表面 5.09 胃壁 5.19 皮膚 3.48 大腸壁 4.01 脾臓 1.09 心臓 8.43 精巣 1.73 腎臓 1.22 胸腺 5.62 肝臓 5.52 甲状腺 4.88 肺 3.75 膀胱 4.53 子宮 5.30 全身の線量当量は 4.30 Gy/MBq 8) 166 13.その他の神経伝達系プローブ合成 [定量解析のための低比放射能製剤調製法] 受容体密度(Bmax)は、高比放射能および低比放射能の S−(−)−[11C]CGP-12177 製剤の 2 回投与を行い、左室心筋および左室内腔の時間放射能曲線を作成し、グラフ解析法を用いて算 出する 9,10)。低比放射能の S−(−)−[11C]CGP-12177 製剤は、次の手順で S−(−)−[11C]CGP-12177 に、非放射性 S−(−)−CGP-12177 を添加して次の手順で調製する。 1. S−(−)−CGP-12177(註 1)を精秤し、注射用生理食塩液を加えて、一定濃度の溶液を 調製した後、無菌ろ過(0.22 μm)する。 2. 滅菌バイアルに、一定量を小分けして、密封し、添加用 S−(−)−CGP-12177 溶液とする。 (註2、3) 3. 短寿命放射性薬剤品質管理基準に適合した S−(−)−[11C]CGP-12177 注射液に一定量の 添加用 S−(−)−CGP-12177 溶液を加え、S−(−)−[11C]CGP-12177 製剤(低比放射能製剤) とする。(註4) 註1) 非放射性 S−(−)−CGP-12177 は、1) 市販のラセミ体(TOCRIS 社、Cat. No:1134、和 光純薬で購入可能)を HPLC で光学分割するか、2)ジアミノ前駆体とトリホスゲン との反応により得られる。 註2) 添加用 S−(−)−CGP-12177 溶液は冷暗所に保存する。 註3) 添加用 S−(−)−CGP-12177 溶液の有効期限は安定性試験の結果により定める。 註4) 低比放射能の S−(−)−[11C]CGP-12177 製剤は薬物として 30~40 μmg(100~140 nmol) の S−(−)−CGP-12177 を含有するため、ヒトへの投与に当っては、通常の PET 薬剤に 関する基準に加えて、薬物量を考慮した基準の設定が必要である。 参考文献 1. Elsinga P.H., Van Waarde A., Vaalburg W.: Eur. J. Pharmacol., 499, 1–13 (2004). 2. Aigbirhio F., Pike V.W., Francotte E., et al.: J. Label. Compd. Radiopharm., 31, 159–161 (1992). 3. Aigbirhio F., Pike V.W., Francotte E., et al.: Tetrahedron Asymm., 3, 539–554 (1992). 4. Boullais A., Crouzel C., Syrota A.: J. Label. Compd. Radiopharm., 23, 565–567 (1986). 5. Brady F., Luthra S.K., Tochon-Danguy H., et al.: Appl. Radiat. Isot., 42, 621–628 (1991). 6. Hammadai A., Crouzel C.: J. Label. Compd. Radiopharm., 29, 681–690 (1991). 7. Nishijima K., Kuge Y., Seki K., et al.: Nucl. Med. Commun., 25, 845–849 (2004). 8. Nishijim K.: Yakugaku Zasshi, 126, 737–745 (2006). 9. Delforge J., Syrota A., Lancon J.P., et al.: J. Nucl. Med., 32, 739–748 (1991). 10. Delforge J.: J. Nucl. Med., 35, 921 (1994). 167 13-6.[11C]カーフェンタニル合成法 (石渡 喜一) カーフェンタニル(carfentanil、4–((1–oxopropyl)–phenylamino)–1–(2–phenylethyl)–4– piperidine-carboxylic acid methyl ester)は µ オピオイド受容体に対し高い親和性・選択性(KD = 0.08 nM、human cortical and thalamic tissues)を示すアゴニストで 1)、µ オピオイド受容 体を測定できる PET 薬剤として Johns Hopkins 大学で開発、臨床利用された。その後、多く の研究機関で臨床利用され、てんかんなどの脳障害、精神疾患、鎮痛発現機序、薬物依存性、 薬物占拠率、心筋や腫瘍の受容体に関する研究が報告され、近年では特に鎮痛に関連した受容 体賦活研究に関心が持たれている 2-5)。 A.合成法 下記の反応スキームにより合成する。 CO211CH3 CO2Na N N COC2H5 11 CH3OTf, NaOH N N COC2H5 DMF, r.t. [使用試薬] [11C]メチルトリフレート デメチルカーフェンタニル Na 塩PharmaSynth 製 無水 DMFAldrich(22705-6) NaOH特級試薬 [方法] 0.1 M NaOH(6 μL)を含むデメチルカーフェンタニル Na 塩の DMF 溶液(1 mg/mL、0.25 mL、0.62 μmol)に(註1)、室温下で He 気流下(30~50 mL/min)[11C]メチルトリフレート を通して捕集する。直ちに反応液に H2O で 2 倍希釈した HPLC 溶離液(1.3 mL)を加えて希 釈し、HPLC により分離精製する。 分離された[11C]カーフェンタニル溶液は、ロータリエバポレーターのフラスコに分取し、溶 媒を除いた後、生理食塩水に溶解する。 (註2)メンブレンフィルターを通して無菌バイアルに 捕集する。(註2) 註1) 前駆体溶液は−20C で保存して1ヶ月程度の使用には影響がなかったが、半年の保存 では収率がかなり低下した。 註2) アスコルビン酸などの安定化剤なしに、少なくとも調製後 2 時間までは[11C]カーフェ ンタニルの分解は認められなかった。 註3) メンブレンフィルターには、脂溶性薬剤の吸着の少ないマイレックス GV フィルターTM (ミリポア)などを使用する。 168 13.その他の神経伝達系プローブ合成 [合成法の特徴と問題点] 本法では、デメチルカーフェンタニル Na 塩に対して NaOH を 1 当量を使用するとき、放射 化学的収率は[11C]メチルトリフレートに対して 50%を越えるが、2 当量では収率は低下する傾 向があり、大過剰では収率が低下する。NaOH がない時の放射化学的収率は数%であった。 一方、Dannals らの方法では、デメチルカーフェンタニル Na 塩を溶かした DMF 溶液に[11C] よう化メチルをトラップして 35℃で 3 分間加熱するが、塩基は使用しない 6)。また、Studenov らは、デメチルカーフェンタニルの Na 塩あるいは NH4 塩に対して少量の tetra–n–butylammonium hydroxide を加え、液相法やループ法で[11C]よう化メチルと反応させた 7)。 [11C]メチルトリフレート法に関しては、Jewett は遊離酸型のデメチルカーフェンタニルの DMSO 溶液に少量の tetra–n–butylammonium hydroxide を加え、これに[11C]メチルトリフレ ートを吹込んで合成している 8)。 [HPLC 分取条件] カラム:YMC-Pack ODS-A(内径 10 mm×長さ 250 mm)、ワイエムシー 溶離液:CH3CN/50 mM AcOH/50 mM AcONH4(35/32.5/32.5) 流 速:5 mL/min 検出器:UV(260 nm)、線検出器 保持時間:原料 3.8 min、目的物 9.3 分 B.分析法 [放射化学的純度] HPLC i) カラム:TSKgel Super-ODS(内径 4.6 mm×長さ 100 mm) 、東ソー 溶離液:CH3CN/50 mM AcOH/50 mM AcONH4(30/35/35) 流 速:1 mL/min 検出器:UV(220 nm)、線検出器 保持時間:原料 1.8 分、目的物 4.5 分 ii) カラム:TSKgel ODS-140HTP(内径 2.1 mm×長さ 50 mm、2.3 m)、東ソー 溶離液:CH3CN/50 mM AcOH/50 mM AcONH4(25/37.5/37.5) 流 速:0.5 mL/min 検出器:UV(220 nm)、線検出器 169 保持時間:原料 1.0 分、目的物 2.5 分 C.その他 [被曝線量]9) ヒト全身 PET 動態計測による。 実効線量:4.6 Sv/MBq 膀胱壁:36.5 Sv/MBq、肝臓:9.7 Sv/MBq、腎臓:4.3 Sv/MBq 全身:2.7 Sv/MBq [急性毒性] LD50:ラット、 7.86 mol(3.2 mg)/ kg(i.v.) 10) [薬理作用] カーフェンタニルはμ-オピオイド受容体に対し高い親和性を有し、モルヒネの 10,000 倍、 フェンタニル(全身麻酔導入時 1.5-8 g/kg を緩徐に静注)の 100 倍の鎮静作用を持つ。動物 には使用されているが、ヒトには使用されていない。10) Tail-withdrawal test(尻尾を熱湯につけ尻尾をはねるまでの潜時をみる尻尾引っ込み試験) による薬理効果は次の通りである。10) Lowest ED50:ラット 1.01 nmol/0.4 g/kg(i.v.) Johns Hopkins 大学では[11C]カーフェンタニルによる臨床研究を開始するに先立ち、麻酔専 門医師により非放射性のカーフェンタニルを用いた single subject toxicity study(マイルドな 効果の現れる 9 mg まで緩徐に静注)を施行し、[11C]カーフェンタニルの最大薬物量をおおよ そ 0.1 g/kg と定め、その後の臨床研究で、副作用等の安全性に関する問題は認められていな い。4) 最近の被曝線量評価の研究では、薬理量以下の 0.03 g/kg(1.8 g/60 kg)で多少の眠気 (primarily mild drowsiness)をきたした被験者おり、その効果が最も大きかった被験者では 呼吸数と血圧がやや低下したが、いかなる臨床的に重要な副作用はなかったと報告された。9) 参考文献 1. Titeler M., Lyon R.A., Kuhar M.J., et al.: Eur. J. Pharmacol., 167, 221–228 (1989). 2. Zubieta J.K., Stohler C.S.: Ann. N.Y. Acad. Sci., 1156, 198–210 (2009). 3. Sprenger T., Berthele A., Platzer S., et al.: Eur. J. Pain, 9, 117–121 (2005). 4. Frost J.J.: Nucl. Med. Biol., 28, 509–513 (2001). 5. Koepp M.J., Duncan J.S.: Adv. Neurol., 83, 135–156 (2000). 6. Dannals R.F., Ravert H.T., Frost J.J., et al.: Int. J. Appl. Radiat. Isot., 36, 303–306 (1985). 7. Studenov, A.R., Jivan, S., Buckley, K.R. et al.: J. Label. Compd. Radiopharm., 46, 837–842 (2003). 8. Jewett D.M.: Nucl. Med. Biol., 28, 733–734 (2001). 170 13.その他の神経伝達系プローブ合成 9. Newberg A.B., Ray R., Scheuermann J., et al.: Nucl. Med. Commun., 30, 314–318 (2009). 10. Chemical Research, Development and Engineering Center (CRDEC-TR-88029). 13-7.[11C]mHED 合成法 [11C]mHED(1R,2S–(–)–[11C]meta–hydroxyephedrine)はノルエピエフリン誘導体であり、 Rosenspire K.C.らにより合成された 1)。[11C]mHED は、交感神経終末における伝達物質であ るノルエピネフリン(NE)と同様、交感神経への特異的取り込み機構である uptake–1 を介し て神経終末に取り込まれ貯留小胞に蓄えられる。しかしながら、その後は NE とは異なり、カ テコール–O–メチル転移酵素(COMT)、モノアミン酸化酵素(MAO)による代謝を受けない。 そのため投与後の放射能分布を測定することにより、交感神経終末機能(主に Uptake–1)を 評価する 2)。また神経内分泌腫瘍の診断薬として臨床研究にも用いられている 3)。 A-1.[11C]よう化メチル法 (西嶋 剣一、久下裕司) 下記の反応スキームにより合成する。 OH OH 11 HO NH 2 CH3I DMSO HO 11 NH CH3 [使用試薬] [11C]よう化メチル Metaraminol(free base)———ABX(註 1) 無水(有機合成用)DMSO –———Aldrich、Wako 註1) ABX 社の Metaraminol (free base)は、購入したものに無水 DMSO を加え、合成 1 回 分(1 mg/0.5 mL)に小分けしている。小分け後 3 カ月程度の使用実績がある。 [方法] Metaraminol free base(1 mg)を無水 DMSO(0.5 mL)に溶解させる(註 1)。この溶液を 反応容器に加え、室温下、N2 気流下(50 mL/min)[11C]よう化メチルを通し 100°C で 5 分間 反応させる。反応終了後、反応液に HPLC 溶離液(1.5 mL)を加えて HPLC により分離精製 する。溶出した目的分画をロータリエバポレーターに導入し、エタノールを減圧除去した後、 生理食塩液に溶かし、最終的に 0.22 μm のメンブレンフィルターを通して注射用薬剤とする。 註1) Rosenspire K.C.ら 1)は、反応溶媒として DMF/DMSO(3/1)混液を用いている。しか しながら逆相 HPLC による分離精製では DMF を除去することができず製剤中への混 入が確認される。 171 [合成法の特徴と問題点] [11C]よう化メチル法および[11C]トリフレート法が報告されている 4)。[11C]よう化メチル法に おいては、立体異性体と考えられる不純物が生成する 1)。分離精製 HPLC において目的物の直 後に立体異性体が溶出されるため分取に注意しなければならない。 [HPLC 分取条件] カラム:Develosil PRAQUEOUS(5 μm、内径 10 mm×長さ 250 mm、ガードカラム: 内径 8 mm×長さ 10 mm)(註 1)、野村化学 溶離液:第一溶出液 6 mM NaH2PO4、第二溶出液 EtOH/H2O(1/99) 流 速:7 mL/min 検出器:UV(254 nm)、放射能検出器 保持時間:第一溶出液により前駆体を溶出後、第二溶出液にて溶出したとき目的物は 18 min 前後に溶出される。 註1) 溶離液として 100%水系を用いているため、カラムの性能によっては保持時間の短縮が 起こり、再現性が得られない。そのため 100%水系においても再現性を有する C30 カ ラム(Develosil PRAQUEOUS)を用いている。 A-2.[11C]メチルトリフレート法 (伊藤 由麿) 下記の反応スキームにより合成する。 [ 11 C]MeOTf HO MeCN 65℃, 2min NH2 HO 11 NH CH 3 [使用試薬] [11C]メチルトリフレート Metaraminol(free base)———ABX 社(3380) (註1) 172 13.その他の神経伝達系プローブ合成 無水アセトニトリル———Merck (12636) 無水メタノール———Ardrich(322415) メイロン(注射用 7%炭酸水素ナトリウム水溶液) 註1) あるいは Sigma の metaramiol bitartrate salt (33402-03-8)から、脱塩して調製できる。 Metaraminol bitartrate(250 mg)を注射用蒸留水(500 μL)に溶解後、重曹(500 mg) を加える。水層に酢酸エチル(50 mL)を加え、溶媒抽出後、有機層を無水硫酸ナト リウムで脱水し、ろ過する。ロータリエバポレーターで酢酸エチルを除去し、得られ た残渣を無水エタノールに溶解し、清浄なバイアルに回収する。これを、アルゴンガ ス加圧下、遮光冷蔵保存する。溶解後 5 週間使用可能である。 [方法] あらかじめ作成しておいた metaraninol /無水メタノール(0.4 mg/50 μL)(註1)に、無水 アセトニトリル(300 μL)を加えた液に、常法で得られた[11C]メチルトリフレートを 30~50 mL/min の流速で通して捕集する。捕集後、65˚C で 2 分間反応させる。反応液に注射用蒸留水 (0.5 mL)を加え(註2)、良く撹拌後 HPLC にて分取する。分取液は、メンブランフィルタ ーを通じてメイロン(30 μL)入り無菌バイアルへ捕集する。 註1) Metaraminol(free base)に無水メタノールを加え、清浄なバイアルにアルゴンガス 加圧下で遮光冷蔵保存する。溶解後 5 週間使用可能。 註2) ここでの水量が少ないと、ピーク形状が異常になる可能性があり、注意が必要。 [HPLC 分取条件] カラム:Megapak SIL C18-10(内径 7.5 mm×長さ 250 mm) 、日本分光(註1)(註2) 溶離液:EtOH/10 mM NaH2PO4(1/99) 流 速:7.0 mL/min 検出器:UV(280 nm)、線検出器 保持容量:90 mL 註1) カラム管理:通常、カラムは消毒用エタノールで満たしておく。使用前に EtOH/10 mM NaH2PO4(1/99)を流し、エタノールを十分除きつつ平衡化する。 尚、当該条件下では、毛管作用によりカラム充填剤細孔から移動相が抜け出て、保 173 持容量が減少する恐れがあるので、平衡化後、使用直前までカラムを若干の加圧状態 にしておく。使用後は、再び消毒用エタノールで置換して保管する。 註2) 当該条件下では、全量を分取すると、mHED の手前に不純物ピークが混入することが ある。この場合、最初の一部分を捨てて分取する必要がある。 [合成法の特徴と問題点] 本法は、よう化メチル法に比べて低い温度で短時間に反応が進行し、収率も高い。本法では、 分取 HPLC に C18 逆相カラム、溶離液に EtOH/10 mM NaH2PO4(1/99)を用いており、カ ラムの劣化が早いが、溶離液をフィルトレーション後、pH 調整して直接注射液と出来る。ただ し、微量のエタノールを含有するので、アルコール過敏症の患者等では禁忌である。 B.分析法 [放射化学的純度] HPLC i) カラム: Xbridge C18 (内径 4.6 mm×長さ 150 mm、5 μm)、Waters 溶離液:CH3CN/ 100 mM HCOO NH4(5/95) 流 速: 1 mL/min 検出器:UV(254 nm)、線検出器 溶出時間:前駆体 3.6 分、目的物 4.7 分前後 ii) カラム:TSKgel Super ODS(内径 4.6 mm X 長さ 100 mm、2.3 μm)、東ソー(註1) 溶離液:EtOH/20 mM NaH2PO4/85% H3PO4(15/500/0.5)pH 2.5 流 速:0.5 mL/min 検出器:UV(280 nm)、線検出器 保持容量:原料 1.8 mL、mHED 2.2 mL 註1) 当該条件下では、毛管作用によりカラム充填剤細孔から移動相が抜け出て、保持が減 少するので、平衡化後、使用直前まで、カラムを若干の加圧状態にしておく。 C.その他 [毒性] 拡張型単回静脈内投与毒性試験 mHED の 0.18 及び 2 mg/kg を 1 群雌雄各 6 匹の Crl:CD (SD) ラットに単回静脈内投与、対 照群(雌雄各 6 匹)には媒体(生理食塩液)を同様の方法で投与した[各群それぞれに投与翌日 剖検群(雌雄各 3 匹)及び投与後 14 日剖検群(雌雄各 3 匹)を設けた]。その結果、被験物質 投与に起因したと考えられる変化は 2 mg/kg 群において肺に認められた。病理組織学的検査に おいて、投与翌日剖検群の雌雄で軽度又は中等度の出血を伴った炎症が巣状に観察された。投 与後 14 日剖検群では雄 1 例で軽度な出血を伴った炎症巣、雌 1 例で炎症巣がみられたが、回 復性が確認された。一般状態、体重、血液学的検査、血液生化学的検査では、毒性学的に意義 のある変動は認められなかった。本試験条件下において、最大無毒性量は 0.18 mg/kg と推察さ 174 13.その他の神経伝達系プローブ合成 れた。なお、2 mg/kg において肺に出血を伴った炎症巣が認められたが、回復性がみられる変 化であった。 [11C]mHED 製剤による単回投与毒性試験 [11C]mHED 製剤(120 MBq/kg: 臨床想定の最大投与量 740 MBq/60 kg の 10 倍量)を雄性 Wistar ラット(WKAH)の尾静脈内に投与、また対照群として生理食塩液を 0.2 mL 投与した (各 1 群 6 匹)。12 日間の体重測定と全身症状観察を実施した結果、いずれの群のラットにも 死亡例はなく、全身症状においても特記すべき変化は見られなかった。体重は、[11C]mHED 製 剤投与群と対照群とに有意な差は見られなかった。 参考文献 1. Rosenspire K.C., Haka M.S., Van Dort M.E., et al.: J. Nucl. Med., 31, 1328–1334 (1990). 2. Schwaiger M., Kalff V., Rosenspire K., et al.: Circulation, 82, 457–64 (1990). 3. Shulkin B.L., Wieland D.M., Schwaiger M., et al.: J. Nucl. Med., 33, 1125–1131 (1992). 4. Någren K., Muller L., Halldin C., et al.: Nucl. Med. Biol., 22, 235–239 (1995). 175
© Copyright 2026 Paperzz