Paper
zz
Explore Categories
Log in
Create new account
No category
Free Full Text ( Final Version , 3mb )
Download
Report
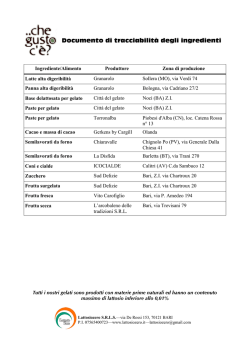
Documento di tracciabilità degli ingredienti
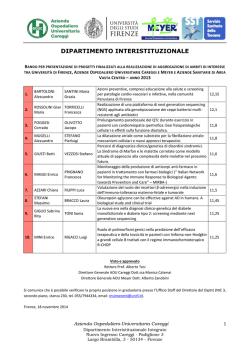
Elenco progetti finanziati - Azienda Ospedaliera Careggi
Seminario Vincenzo Nigro - Università degli Studi di Torino
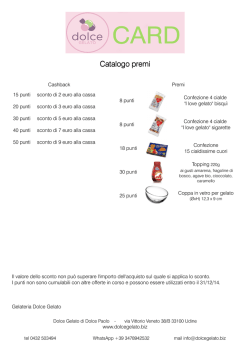
Catalogo premi - Dolce Gelato
LABO-XP
Basic Next Generation Sequencing (NGS) procedures
L`AIFA pubblica il nuovo Algoritmo per la terapia dell`Epatite C cronica
Menu del Salad Bar
Leggi tutto - IPSEOA "V. TITONE"
apri o scarica
© Copyright 2026 Paperzz
About Paperzz
DMCA / GDPR
Report