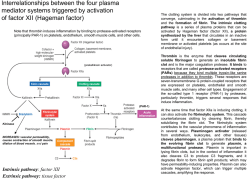
MALATTIE DEL SANGUE Prof Di Raimondo Lezione 1 Il prof farà delle lezioni per introdurci all’ematologia. Non è che chiederà agli esami quello che ha detto a lezione. Quindi dobbiamo studiare sul libro e non sulle sue lezioni. Le sue lezioni servono a darci una chiave di lettura di quello che c’è scritto sul libro. Cominciamo con qualche ricordo di biologia e citologia. SANGUE: FUNZIONE E COMPOSIZIONE Nel sangue noi abbiamo una parte liquida che è il plasma e una parte corpuscolata formata dai globuli rossi, dai globuli bianchi e dalle piastrine. I globuli rossi sono gli elementi a disco biconcavo, le piastrine sono dei piccoli organelli e i globuli bianchi sono rappresentati dal loro polimorfismo. Il sangue è fondamentalmente un tessuto connettivo che serve a trasportare e a proteggere. Che cosa trasporta? Trasporta ossigeno, trasporta CO2, acqua, vari nutrienti, ormoni. La sua funzione di protezione è esplicata nei confronti della capacità di coagulazione, dell’infiammazione e del sistema immunitario. E poi è molto importante anche per regolare il pH e la temperatura. Si dice che gli adulti hanno circa 5 litri di sangue, che varia ovviamente a seconda della superficie corporea, e di questi 5 litri di sangue il 55% è costituito da plasma, e il resto è formato da cellule, soprattutto globuli rossi. Il sangue ha anche delle proprietà fisiche, quali la viscosità o l’osmolarità, e se per esempio abbiamo un’alta osmolarità o viscosità ci possono essere dei problemi di pressione arteriosa. Come si diceva prima esiste una parte corpuscolata formata prevalentemente da globuli rossi, ma anche da globuli bianchi e da piastrine e questa parte corpuscolata costituisce circa il 45% di tutto il sangue, per cui se voi prendete un pò di sangue, lo mettete in un capillare e lo centrifugate otterrete in questa microprovetta questa differenza di distribuzione: il 45% è rappresentato da elementi corpuscolati,di cui la maggior parte i globuli rossi, e questo è quello che forma l’ ematocrito, mentre il 55% è formato dal plasma. Nel plasma c’è acqua, proteine, in particolare l’albumina, sali, composti organici ecc. ci sono prodotti di degradazione, la bilirubina, l’acido urico, ci sono i gas, ci sono gli elettroliti, tutto questo serve a far funzionare l’organismo. Fra le proteine più importanti c’è l’albumina: ricordiamo che viene prodotta dal fegato, è la proteina più abbondante tra le proteine plasmatiche e che viene a contribuire in maniera significativa alla viscosità e all’osmolarità del sangue, e quindi influenza sia la pressione del sangue sia il bilancio elettrolitico, ricordate che il paziente con impoalbuminemia è un paziente con degli edemi molto importanti, cosiddetti anasarcatici. Un’altra proteina molto importante e presente nel plasma è il fibrinogeno assieme a tutti i fattori della coagulazione: anche questi sono quasi tutti prodotti dal fegato, e il fibrinogeno, che poi è la molecola a valle della cascata coagulativa, viene convertito in fibrina che serve a formare il coagulo. Oltre a questi ci sono le globuline che sono prodotte dalle immunocellule che sono uno dei pilastri più importanti del sistema immunitario. EMOPOIESI Si intende per tessuto emopoietico il tessuto che produce i globuli bianchi, i rossi e le piastrine e che subisce un’evoluzione dalla vita embrionale alla vita adulta. Inizialmente è contenuto nel sacco vitellino, poi nel fegato e infine dalla nascita in poi è contenuto quasi esclusivamente nel midollo osseo. Nel midollo osseo ci stanno le cosiddette cellule staminali che daranno origine a quello che noi troviamo nel sangue periferico. Le cellule staminali sono delle cellule in grado di differenziarsi verso le varie linee cellulari, ma in grado anche di autoriprodursi. Oltre al sistema linfoide, che è rappresentato dal timo, dai linfonodi, dalle tonsille e dalla milza, e che serve non soltanto alla produzione, ma soprattutto alla maturazione di quello stipite cellulare che è lo stipite linfoide. Ricordatevi sempre che la produzione di globuli rossi è regolata da un ormone che è l’eritropoietina che viene prodotta dal rene, in piccola quota anche dal fegato. Vediamo la schematizzazione di quello che succede nel midollo osseo dove dalla cellula staminale si diramano diverse cellule cosiddette commissionate che daranno origine o allo stipite linfoide, i progenitori dei T linfociti e B linfociti o allo stipite mieloide propriamente detto che darà origine poi nel sangue periferico a neutrofili, basofili, eosinofili e monociti, allo stipite megacariocitico, che darà origine alle piastrine e allo stipite eritroide che darà origine agli eritrociti. Quindi questi elementi che noi troviamo nel sangue periferico normalmente, non sono altro che il frutto di una sorta di albero maturativo che parte dalla cellula staminale e che attraverso un complicato sistema di controllo e di maturazione, attraverso una serie di citochine e di fattori trascrizionali, determina il passaggio e la maturazione verso le varie linee cellulari. Queste cellule stanno nel midollo osseo che è un tessuto complesso: il midollo osseo è quello che sta dentro le ossa (immagine: frammento di osso decalcificato che fa vedere le trabecole ossee all’interno delle quali è contenuto il midollo osseo). Però questo midollo osseo prende rapporti con questo tessuto osseo, dove ci stanno non solo matrice ossea, ma anche gli osteoblasti e gli osteoclasti, voi sapete che gli osteoblasti e gli osteoclasti sono cellule che regolano la funzione ossea: gli osteoclasti servono a riassorbire l’osso, gli osteoblasti a riprodurre osso. E nel midollo osseo non ci stanno soltanto cellule emopoietiche, la cellula staminale e tutte le sue figlie, ma ci stanno anche vasi, cellule stromali, adiposità, fibrobllasti, macrofagi, mastociti, la matrice extracellulare, tutti questi elementi costituiscono il cosiddetto microambiente midollare, che è l’ambiente dove il tessuto emopoietico cresce e si divide. E quindi è molto importante che queste cellule prendano rapporti con le cellule stromali, si dice che esistono proprio delle nicchie di tessuto emopoietico, formate da diverse cellule stromali in cui entrano in gioco non soltanto i fibroblasti, le cellule mesenchimali, ma anche gli osteoblasti e gli osteoclasti e il controllo consiste in una sorta di interazione tra queste cellule emopoietiche e cellule stromali che si “parlano” tra di loro e questo serve poi alle cellule emopoietiche a produrre la cellule figlie che attraverso diverse maturazioni, diverse divisioni e sotto il controllo di diverse citochine daranno luogo alle cellule commissionate, ai progenitori e infine agli elementi maturi. Quindi noi dobbiamo considerare che esistono questi due compartimenti, il midollo e il sangue periferico che sono in continuo equilibrio fra di loro: nel midollo dobbiamo considerare che esiste una componente staminale, la quale può anche andare in circolo ( è stato dimostrato che queste cellule staminali possono passare in circolo), ma che comunque prevalentemente sta nel midollo in determinate nicchie emopoietiche, queste cellule staminali daranno poi origine alla linea granulocitica, alla linea eritroide e alla linea megacariocitica. Immagine: gli elementi pluripotenti sono rappresentati da questo elemento chiaro, gli elementi commissionati da questo elemento più scuro, e gli elementi proliferativi, cioè il grosso dell’esercito che rappresenta questa proliferazione midollare è rappresentato dagli elementi che proliferano e maturano, e come si vede c’è una differenza tra il comparto megariocitico, quello eritroide e quello granulocitico, nel senso che nel midollo osseo non sono presenti soltanto gli elementi immaturi della serie granulocitaria ma sono presenti anche elementi più maturi, elementi che poi riscontriamo nel sangue periferico, i granulociti per intenderci, ce ne sono tanti nel midollo perché costituiscono qui un pool di riserva; questo pool di riserva invece non è presente nel midollo per quanto riguarda gli elementi eritroidi e per quanto riguarda gli elementi megariocitici. Lo stesso nel sangue periferico non sono presenti soltanto circolanti, ma sono presenti anche elementi che sono in qualche modo marginati e che servono anche lì come elementi di riserva, pool di riserva, e sono presenti sia per quanto riguarda i granulociti che per quanto riguarda gli ??. Vediamo come funziona questa emopoiesi. Parliamo al momento solo della produzione degli eritrociti. Abbiamo già detto che l’eritropoiesi si modifica nella vita embrionale dal sacco vitellino passa alla vita fetale nel fegato e quindi successivamente nella vita adulta inizia la produzione midollare e nella vita adulta diciamo che è soltanto midollare, midollare nel senso di midollo osseo presente all’interno delle ossa e nei bambini è presente anche all’interno delle ossa lunghe, nell’adulto è presente soltanto a livello delle ossa piatte e nello scheletro assiale, cioè bacino, colonna vertebrale, anche alcune coste, mentre invece nel bambino lo troviamo anche nelle ossa lunghe, nel femore, nell’omero. Immagine: sezione di midollo osseo dove si vede nettamente la matrice ossea e poi tutto il tessuto midollare, il tessuto midollare è inframezzato da queste lacune che non sono altro che lacune di grasso, il cosiddetto midollo adiposo, si riconoscono molto facilmente i magacariociti che sono cellule molto grandi e si vedono anche a piccolo ingrandimento. Come viene controllata questa eritropoiesi? Vediamo di nuovo questa sorta di albero che dalla cellula staminale dà origine alle cellule più mature e in realtà noi non riusciamo a seguire su un piano morfologico tutta questo albero perché non sappiamo com’è fatta sul piano morfologico la cellula staminale, come non sappiamo com’è fatta sul piano morfologico la cellula immediatamente successiva la cellula staminale, però sappiamo che queste cellule esistono sul piano funzionale tanto che vengono chiamate CFU (colony forming unit), cioè delle cellule in grado, in determinate condizioni di coltura, di formare le colonie che daranno origine a nuove cellule che sviluppandosi formano una colonia, e esistono le cosiddette CFUs (spleen, perché è stato dimostrato che nel topo si trovano nella milza) e queste daranno origine a tutta la linea mieloide cosiddetta, che darà origine alla linea megacariocitica, alla linea mieloide propriamente detta e alla linea monocitaria; da questi si dipartono anche la linea che andrà a formare il globulo rosso e che prevede una tappa intermedia funzionale che viene definita BFU-E cioè burst forming unit-erythroid, che non sono altro che delle colonie un po’ più piccole, sono delle blast forming unit eritroidi. Queste sono due tappe funzionali del processo maturativo dell’eritrone che darò origine poi al globulo rosso. Quindi:dalle cellule staminali -> CFU->BFU-E . Quindi tutto comincia da una singola cellula, da un cellula staminale che qualcuno chiama emocitoblasta, cellula staminale che peraltro costituisce un elemento comune che poi darà origine non soltanto alla linea emopoietica ma anche alle cellule endoteliali, sia le cellule del sangue che le cellule endoteliali derivano da un progenitore comune. Il globulo rosso che origina non è altro che un contenitore di emoglobina e come avviene questo processo? Da questa cellula così complessa, con nucleo, apparato di golgi, mitocondri ecc., si deve formare questo contenitore, questo globulo rosso che non è altro che una cellula priva di nucleo che serve appunto soltanto da contenitore, quindi non ha tutte queste funzioni esplicate da una qualunque cellula. Quindi sappiamo dove comincia la sintesi dell’emoglobina perché sappiamo riconoscere anche sul piano morfologico questi elementi che chiamiamo proeritroblasti, poi eritroblasto basofilo. Questi sono gli elementi che noi sappiamo riconoscere al microscopio, perché gli elementi più immaturi tra cui la cellula staminale, le BFU-E non sappiamo che faccia hanno, li cominciamo a riconoscere soltanto a partire dal proeritroblasto, cioè dalla tappa successiva a quelle delle BFU-E. Quindi da questa cellula, dall’eritroblasto basofilo comincia la sintesi di emoglobina. Perché si chiama eritroblasto basofilo? Perché produce tanto RNA e questo RNA si colora essendo un acido con un colorante basico e quindi basofilo. E quindi noi vediamo questo citoplasma blu che indica che c’è tanto RNA in questa cellula. Successivamente comincia la sintesi dell’eme, perché in questa cellula sono presenti dei recettori per la trasferrina che gli trasporta il ferro all’interno della cellula e quindi si comincia a vedere la prima emoglobina, per cui a quel colore blu che vedevamo perché appunto c’è tanto RNA si aggiunge adesso un colore rosso che è quello dell’emoglobina per cui questo eritroblasto, da che è basofilo diventa policromatofilo perché ha questa policromasia legata al fatto che il citoplasma non è più blu per il tanto RNA ma diventa blu mischiato con rosso perché inizia ad essere sintetizzata l’emoglobina. Quando la sintesi dell’emoglobina è completa abbiamo il cosiddetto eritroblasto ortocromatico, significa che ha lo stesso colore del globulo rosso, perché appunto contiene nel suo citoplasma tanta emoglobina. A questo punto per accumulare più emoglobina l’ortocromatico si libera del nucleo, e quindi c’è un processo proprio di espulsione del nucleo, che si vede anche al microscopio elettronico, rimanendo poi soltanto il citoplasma che poi costituirà il globulo rosso. Prima di diventare globulo rosso l’eritroblasto ortocromatico si chiama reticolocita, che è la tappa immediatamente precedente quella del globulo rosso, e si chiama reticolocita perché in questa cellula che non ha più nucleo, sono presenti dei residui di RNA e questi residui si colorano con una colorazione che è il cristal violetto, e che dà questo aspetto reticolare, e quindi viene chiamato reticolocita. Infine la tappa maturativa finale è quella del globulo rosso. Allora dall’emocitoblasta, cioè la cellula staminale (qualcuno lo chiama anche emoangioblasta perché da origine all’endotelio o comunque alla linea diciamo vascolare) → si passa al pro eritroblasto (eritroblasto basofilo), quello molto ricco di RNA e proteine che produce appunto l’emoglobina → il policromatofilo, dove questa emoglobina comincia a vedersi → l’ortocromatico dove c’è tutta emoglobina → il reticolocita che non ha il nucleo ma ha ancora i residui di RNA → e infine l’eritrocita. Vediamo un’immagine: rappresentazione grafica di come cambiano i colori, si vede la cellula staminale iniziale… vediamo come il nucleo progressivamente si riduce, diventa picnotico e alla fine viene espulso e il colore cambia da un’intensa basofilia ad una policromasia fino al colore ortocromatico. Quindi per riassumere l’eritropoiesi, questa viene prodotta dalla cellula staminale del midollo osseo, qui subisce un processo di maturazione progressiva fino a dare luogo ai reticolociti, i reticolociti poi passano nel sangue e dopo loro ulteriore maturazione diventano eritrociti; qui viene anche rappresentata la durata di questo processo, grosso modo 6 giorni per fare maturare un proeritroblasto, 2 giorni per far maturare un reticolocita e poi 120 giorni per fare diventare un nuovo eritrocita vecchio eritrocita. In tutto questo processo di maturazione intervengono tutta una serie di fattori trascrizionali molto complessi che vengono attivati e inattivati a seconda delle varie tappe maturative. In questa maturazione un ruolo estremamente importante lo giocano i macrofagi. Al centro dell’isolotto eritropoietico sta una cellula macrofagica e tutt’intorno ci stanno questi elementi eritroidi, questo perché in questo processo maturativo non c’è soltanto questa cellula che si modifica progressivamente ma ci sono tutta una serie di fattori, di citochine comprese le cellule stromali, è importante il contatto dell’eritrone con questi elementi che nell’insieme costituiscono il microambiente midollare. Uno dei meccanismi di regolazione di questo processo è un meccanismo a feedback dove, è stato dimostrato abbastanza recentemente, gli eritroblasti più maturi agiscono in maniera inibitoria sugli eritroblasti più immaturi attraverso il sistema del FAS-Fas ligand. Gli eritroblasti più immaturi presentano sulla loro superficie questo recettore Fas che viene definito un recettore di morte, cioè l’attivazione di questo recettore porta a morte la cellula. È stato dimostrato che gli eritroblasti più maturi presentano sulla loro superficie il ligando del Fas, che è un meccanismo di controllo della produzione: quando ci sono tanti elementi maturi questi inibiscono gli elementi immaturi attraverso la stimolazione del recettore Fas con l’induzione dell’apoptosi. Sono importanti i macrofagi che hanno un ruolo duale, cioè da un lato i macrofagi servono come cellule nutrici per l’elemento eritroide, la loro presenza e funzione è estremamente importante per far maturare questi elementi eritroidi, dall’altro lato i macrofagi poi servono a fagocitare il globulo rosso senescente, quindi esistono macrofagi che favoriscono lo sviluppo del globulo rosso maturo e macrofagi che invece servono a eliminare i globuli rossi senescenti. Questi globuli rossi una volta formati a che servono? Servono fondamentalmente a trasportare l’O2 ai tessuti, a trasportare la CO2 indietro ai polmoni e sono delle cellule che hanno tipicamente una forma a disco biconcavo con un diametro di 7,5 µ, senza organelli, non hanno più mitocondri, non hanno più RNA, è proprio un contenitore per l’emoglobina, sono fatti in modo tale da sfruttare il più possibile la loro superficie per la loro funzione più importante che è quella dello scambio dell’ossigeno. Quindi hanno un perfetto design per lo scambio dei gas. Ma hanno un design perfetto anche per attraversare i capillari, è importante che questi globuli rossi siano abbastanza deformabili per poter attraversare i piccoli vasi, se questo non succede si formano i cosiddetti rouleaux, cioè degli impilamenti di questi globuli rossi, che formano delle ostruzioni vascolari, quindi la deformabilità del globulo rosso è la caratteristica essenziale indispensabile per la sua funzione. Non sempre i globuli rossi hanno lo stesso volume, la stessa forma: - quando non hanno lo stesso volume si parla di anisocitosi; - quando non hanno la stessa forma si parla di poichilocitosi. Vedremo poi l’importanza di questa alterazione dei globuli rossi. Usualmente c’è una aniso-poichilocitosi, di solito quando c’è una variazione della forma c’è anche una variazione del volume. Si dice che i globuli rossi hanno una vita di 120 giorni ed è stato calcolato che addirittura durante questi 120 giorni percorre oltre 700 miglia, non si sa bene come hanno fatto a calcolarlo però per dire come questi globuli rossi costituiscono una vera e propria macchina, e come tutte le macchine ha bisogno della revisione, e durante questo periodo di 120 giorni si passa da un globulo rosso immaturo ad un globulo rosso adulto, fino ad un globulo rosso senescente; il globulo rosso senescente è quello dove c’è una contrazione di volume e una contrazione della sua estensione, fondamentalmente c’è anche una contrazione della sua deformabilità, questa macchina che ha bisogno della revisione ha bisogno di fare il pit-stop, e questo viene fatto nella milza e nel rene, che sono i due organi che controllano il numero e l’efficienza di questa macchina, il globulo rosso è quindi soggetto a dei controlli che avvengono nel rene e nella milza. Nel passaggio attraverso il rene, dalla via arteriosa alla via venosa, vanno a sensibilizzare alcune strutture peritubulari, ed è stato dimostrato che un rene ipoperfuso produce più eritropoioetina. Questi globuli rossi attraversano i capillari e vanno ad essere captati da dei veri e propri sensori dell’ossigeno che sono presenti sulla parete di questi capillari: se ci sono pochi globuli rossi questo recettore per l’ossigeno attiva un sistema che viene regolato dall’ ipoxid induced factor e questo determina l’attivazione del gene che traduce per l’eritropoietina e quindi le cellule peritubulari del rene cominciano a produrre eritropoietina. L’eritropoietina che viene prodotta è in grado di stimolare non tutto l’eritrone ma è in grado di stimolare soprattutto le BFU-E e le CFU-E, agisce molto poco sugli elementi più immaturi commissionati quali il proeritroblasto… agisce prevalentemente sulle bfu-e e sulle CFU-E. Peraltro questa fabbrica è una fabbrica prodigiosa perché riesce a produrre una quantità di globuli rossi inimmaginabile, è stato calcolato che il midollo osseo normale riesce a produrre fino a tre milioni di globuli rossi al secondo. Quando c’è eritropoieina questa riesce a decuplicare la produzione di globuli rossi passando da tre milioni al sec a trentamilioni al sec, quindi è un sistema estremamente efficiente che riesce a potenziare di molto la sua capacità grazie all’eritropoietina. Quando ci sono invece molti globuli rossi, anche qui vengono a contatto con il sensore dell’ossigeno che quindi in questo caso viene spento e si blocca la produzione di eritropoietina. Tutto quindi è legato a questo sensore dell’ossigeno presente a livello delle cellule tubulari del rene. Abbiamo visto l’importanza del rene nell’eritropoiesi. Vediamo invece l’importanza della milza nella regolazione. Se ogni secondo vengono prodotti circe tre milioni di globuli rossi, chiaramente ogni secondo altrettanti devono essere eliminati. Quindi qualcuno li deve togliere e questo compito è affidato alla milza in generale al sistema reticoloendoteliale, prevalentemente presente nella milza, anche nel fegato e nel midollo osseo, ma la funzione più importante è quella della milza. E la milza cosa fa? Controlla la funzionalità della membrana dell’eritrocita che è costituita da un doppio strato di fosfolipidi e proteine che servono allo scambio dei gas ma servono anche a mantenere l’integrità della forma e della deformabilità del globulo rosso. Esistono tutta una serie di proteine che costituiscono questo scheletro, questa impalcatura della membrana eritrocitaria che sono variamente deformabili e le più importanti proteine sono la spectrina e la anchirina. Quindi un’alterazione di queste proteine presenti nella membrana dell’eritrocita, nei confronti di questi si induce una risposta immunologica. La membrana eritrocitaria invecchiata determina una sorta di riconoscimento di questa alterazione: il globulo rosso quando invecchia subisce delle modificazioni, questo viene riconosciuto dal sistema reticoloendoteliale il quale sviluppa degli anticorpi contro questa alterazione e questo determina la fagocitosi del globulo rosso che viene così fagocitato dalla cellula macrofagica e distrutto. Quindi nella milza avviene i riconoscimento degli elementi senescenti o degli elementi che hanno perso la deformabilità, quindi il globulo rosso entra nei sinusoidi della milza e qui subisce il crash test, esattamente come le macchine. Il globulo rosso passa attraverso questi sinusoidi e questi però sono regolati in modo tale che si stringano per cui se il globulo rosso è deformabile e passa il crash test esce di nuovo dalla milza, ma se il globulo rosso non è deformabile e non passa questo esame viene eliminato dalle cellule macrofagiche, quindi nella milza esiste un controllo di quello che è la funzionalità e la deformabilità del globulo rosso, diciamo la milza perché è prevalentemente nella milza, poi ovviamente dobbiamo parlare di sistema reticoloendoteliale. Quindi di globuli rossi se ne producono in quantità enormi, 2-3 milioni al secondo, il prodotto finale di quella catena che abbiamo prima visto possiamo considerare il reticolocita, che è appunto una globulo rosso che presenta ancora dei residui di RNA e che è in grado di passare nel sangue periferico. Quando andiamo a contare questi reticolociti essi sono circa l’1% dei globuli rossi. Il globulo rosso viene regolato dall’eritropoietina e tutte quelle condizioni che determinano un’ipossia sono uno stimolo alla produzione dei globuli rossi per cui in alcune condizioni noi possiamo trovare un aumento dei globuli rossi, per esempio pazienti affetti da insufficienza respiratoria importante hanno una condizione di ipossemia e questa determina un aumento dei globuli rossi per cui questi pazienti sono spesso poliglobulici, hanno un numero elevato di globuli rossi, sono pazienti pletorici, rossi, ovviamente la stessa situazione vale per il fumo, per le alte altitudini dove l’aria è più rarefatta con una carenza di ossigeno e lì aumenta la produzione di eritropoietina, di fatti gli atleti di una volta per ottenere delle prestazioni migliori andavano in altura, adesso fanno direttamente introduzione di eritropoietina. Per produrre globuli rossi ci vogliono alcune sostanze, le più importanti sono: il ferro, la B12 e l’acido folico che riescono a produrre l’emoglobina. Questa è una proteina formata dalla globina più l’eme e ogni molecola di emoglobina può trasportare 4 atomi di ossigeno, l’emoglobina fetale ha una maggiore affinità per l’ossigeno. Dicevamo che l vita del globulo rosso è di 120 giorni e che il globulo rosso senescente perde la deformabilità della membrana e quindi viene distrutto dalla milza, questi macrofagi nella milza o in altre sedi non fanno altro che digerire la membrana del globulo rosso, separare l’eme dall’emoglobina, idrolizzare la globina, cioè farla tornare come aminoacidi che sono quindi nuovamente disponibili, rimuovere il ferro dall’eme e convertire l’eme in blirubina che è indiretta, poi viene coniugata e diventa bilirubina diretta e viene escreta dalla bile con le feci, quindi la degradazione del globulo rosso porta da un lato alla degradazione della globina e quindi alla liberazione di aminoacidi e dall’altro la produzione di bilirubina che viene escreta. È ovvio che quando noi abbiamo un eccesso di distruzione dei globuli rossi uno dei segnali che possiamo evidenziare è l’aumento della bilirubina indiretta. MIELOPOIESI Ovvero la produzione di globuli bianchi. I globuli bianchi sono uno dei pilastri più importanti del s. immunitario. Tra i globuli bianchi distinguiamo i granulociti, neutrofili eosinofili e basofili e gli elementi agranulati che sono i linfociti e i monociti. Sono molti importanti come difesa antibatterica perché sono in grado di fagocitare e di distruggere i germi. I neutrofili costituiscono circa il 60-70% dei globuli bianchi e presentano dei granuli abbastanza ben visibili al microscopio elettronico che vengono distinti in specifici e azzurrofili. Gli eosinofili sono molto di meno, sono intorno al 2-4 %, caratterizzati da questo nucleo bilobato e dalla presenza di granuli peculiari che sono molto importanti sia per combattere parassitosi sia per le risposte allergiche per la presenza di istamina. Poi i basofili sono molto più rari, meno dell’1%, e sono caratterizzati da grossi granuli basofili che contengono molta istamina e quindi sono dei mediatori dell’infiammazione. I linfociti sono estremamente importanti come cellule di difesa immunologica e costituiscono il 30% dei globuli bianchi. E infine ci sono i monociti che costituiscono il 5 % dei globuli bianchi, anche questi sono estremamente importanti nella difesa immunologica. Anche queste hanno origine dalla cellula staminale che darà origine alle cellule dello stipite linfoide e alle cellule dello stipite mieloide. Tutto avviene sempre a livello del midollo. Il midollo conserva e rilascia granulociti e monociti e alcuni linfociti lasciano il midollo per andare a maturare nel timo, e queste sono i linfociti T. Abbiamo visto che i globuli rossi durano 120 giorni, i globuli bianchi, per lo meno i granulociti durano molto meno, circa 5 giorni, i monociti durano poco ma si possono trasformare al di fuori del torrente circolatorio in cellule macrofagiche e lì durano diversi anni. Quindi la maturazione di queste cellule avviene sempre a partire dalla cellula staminale che poi da origine a CFU-M (serie monocita ria), CFU-G (serie neutrofila) e da questo momento in poi li riconosciamo sul piano morfologico e quindi li chiamiamo mieloblasti, promielociti, mielociti. Passano poi nel sangue periferico, ma ricordiamo che esiste un pool di deposito a livello midollare ed esiste un pool di riserva marginale adeso alle pareti dei vasi. Ci vogliono diverse ore per queste diverse tappe ma la cosa importante è che arrivati allo stadio di meta mielociti queste cellule non sono più in grado di maturare mentre invece gli elementi più a monte costituiscono il pool mitotico ed è stato calcolato che ci vogliono dagli 8 ai 12 giorni per avere un neutrofilo; questi vive circa 5 giorni però in realtà è difficile valutare la sopravvivenza perché il neutrofilo può essere compreso in un compartimento circolante oppure in un compartimento marginale: il compartimento circolante circola col sangue, quello marginale è quello che sta adeso alle pareti del vaso e viene utilizzato soltanto nei momenti di bisogno, perché questi globuli bianchi passano dal midollo al sangue periferico e da questo passano necessiti dove servono a combattere le infiammazioni, quindi nel sangue periferico sta per un periodo abbastanza breve e poi passa nei tessuti dove lì poi esplica la sua funzione. Quando in un tessuto c’è un processo infiammatorio, questo richiama i granulociti neutrofili dal sangue dove quindi stanno per molto meno tempo, vanno invece di più nella sede del processo. In questa sede non ci arrivano soltanto i g b che stanno in circolo ma vengono richiamati anche le riserve presenti: quindi i marginati ma anche quelli della riserva midollare. Tutti questi vanno a finire nei tessuti infiammati. La riserva midollare viene quindi mobilitata da diverse condizioni, per esempio le citochine possono mobilizzare la riserva midollare, l’adrenalina può mobilizzare la riserva midollare e altri farmaci alcuni diuretici possono mobilizzare la riserva midollare, per far passare questi neutrofili dal midollo nel sangue periferico. Quindi la cinetica dei neutrofili è rappresentata da questi quadrati su cui agiscono diversi fattori di crescita: GSF, IL-3... sono tutti fattori che servono a far maturare questi elementi dalla cellula staminale verso i granulociti necrofili i quali poi sono distinti in un pool di riserva e un pool marginale. Le piastrine sono prodotte sempre nel midollo a partire da cellule giganti, i megacariociti, che servono alla coagulazione del sangue ma anche alla riparazione della parete dei vasi sanguigni: anche qui tutto ha origine dalla cellula staminale che da origine ai precursori delle piastrine. Vediamo rappresentato un megacariocita e si vede quanto è grande rispetto alle altre cellule. Quindi quando facciamo un prelievo di midollo e andiamo a vedere qual è la popolazione presente in questo midollo possiamo definire il mielogramma. Il mielogramma non è altro che la conta delle cellule che stanno nel midollo. Nel midollo normalmente ci stanno le cellule della linea mieloide, della linea eritroide, i linfociti e i magacariociti che però non entrano nella conta percentuale delle cellule del midollo, quindi non entrano a far parte del mielogramma, si vedono già a piccolo ingrandimento e non vengono contate. Quelli che vengono contati in percentuale sono gli elementi della serie mieloide quindi i mieloblasti, i promielociti, mielociti e gli elementi più maturi, gli elementi della serie eritroide, i linfociti e i monociti. Spesso il prelievo del midollo viene fatto per andare a vedere se ci sono problemi a livello cellulare. Quindi possiamo vedere un midollo normocellulare con un rapporto leuco-eritrocitario alto, cioè ci sono molti più elementi della linea mieloide che della linea eritroide, in genere il rapporto è 2-3/1; possiamo vedere un midollo, di un pzt che ha iperplasia della linea eritroide, che al contrario un rapporto leuco-eritrocitario più basso. Ricordiamo sempre che tutti questi elementi che servono alla mielopoiesi sono sempre in stretto contatto con le cellule stromali, con le cellule endoteliali, con gli adipociti, con macrofagi, cioè c’è sempre un microambiente che serve a poter condurre questa maturazione, compresa quella dei megacariociti. Le cellule staminali hanno sicuramente delle capacità di rigenerazione che sono estremamente importanti: è una cellula indifferenziata in grado di proliferare estesamente, in grado di generare cellule uguali a se stesse per periodi molto lunghi e poi di produrre tutta una serie di cellule differenziate e sapete che c’è una ricca letteratura sulle cellule staminali, tutti ne parlano, pochi le conoscono. Lezione 2 Le cellule staminali sono divise in cellule totipotenti (rappresentate dallo zigote), che sono in grado di dare origine a qualunque cellula dell'organismo, oppure cellule pluripotenti, che sono in grado di differenziarsi in qualunque cellula dell'organismo ma non, ovviamente, negli annessi embrionali (cellule staminali embrionali). E infine abbiamo le cellule staminali adulte che sono prodotte, invece, con la caratteristica di avere una multipotenza, cioè la capacità di generare diversi tipi cellulari all'interno di un tessuto. In ogni tessuto ci sono delle cellule staminali specifiche di quel tessuto. Quindi, per definizione, la cellula staminale è una cellula indifferenziata, è capace di proliferare, è capace di generare delle cellule che sono uguali a se stessa. E questo deve essere fatto per un lungo periodo di tempo in modo tale che queste cellule posso mantenere costante questo compartimento staminale: questo viene definito automantenimento. Ma chiaramente, oltre ad automantenersi, questa cellula staminale deve produrre un numero molto elevato di cellule differenziate: questa è la multipotenzialità. Una caratteristica molto importante di queste cellule staminali è la loro capacità di riparare i danni di quel tessuto. Quindi, fondamentalmente, le cellule staminali servono ad assicurare l'omeostasi del tessuto d'appartenenza e quindi regolano la rigenerazione del tessuto rimpiazzando gli elementi persi per morte naturale o in seguito a ferite o a danni del tessuto. Queste cellule risiedono in determinati microambienti, che si chiamano nicchie, le quali variano in funzione del tessuto d'appartenenza. Noi, in particolare, ci occupiamo delle nicchie delle cellule staminali emopoietiche. All’interno della nicchia la sopravvivenza, l’attività delle cellule staminali è sicuramente condizionata da tutta una serie di condizionamenti sia di tipo umorale, sia di tipo cellulare. Cosa fa la nicchia? - fornisce un microambiente protetto, perché impedisce alle cellule staminali ospiti di venire in contatto con stimoli che servono all'induzione, al differenziamento o servono all'apoptosi; - regola la proliferazione degli elementi staminali, perché una cellula che cresce in maniera incontrollata è come se fosse una cellula neoplastica e quindi si impedisce la formazione di tumori. L'ipotesi è proprio che la differenza tra le cellule staminali normali e le cellule cancerose dipende proprio dal fatto che queste cellule cancerose perdono questo meccanismo inibitorio che viene esercitato, invece, sulle cellule staminali. Nella nicchia emopoietica sono molto ben rappresentati gli osteoblasti, perché il midollo osseo sta nelle ossa. Nelle ossa ci stanno gli osteoblasti e gli osteoclasti, oltre a tante altre cellule, ma sicuramente è dimostrato che gli osteoblasti costituiscono una parte fondamentale della nicchia delle cellule staminali emopoietiche. Le cellule staminali prendono rapporto non solo con gli osteoblasti, ma anche con le cellule stromali, con i vasi sanguigni, con gli elementi che vengono prodotti dalle stesse cellule staminali (gli elementi emopoietici). Le cellule staminali, come abbiamo detto, non hanno una definizione morfologica, ma hanno sicuramente una definizione antigenica, cioè presentano degli antigeni che ci permettono di definire la sottopopolazione di cellule come cellule staminali. In particolare, questi antigeni sono i CD34, che noi utilizziamo molto nella definizione di cellula staminale. La cellula staminale emopoietica, per definizione, è CD34 positiva. Non è che tutte le CD34 positive sono cellule staminali e neanche tutte le cellule staminali sono CD34 positive, però marcando l'anticorpo monoclonale anti-CD34 raccogliamo la maggior parte delle cellule staminali. Inoltre queste cellule staminali presentano una positività per il c-kit, una positività per il CD133 che è in comune con le cellule endoteliali e poi, cosa importante, queste cellule staminali presentano una positività per le pompe che servono a buttare fuori i farmaci. Non solo i farmaci ma anche altre sostanze tossiche per la cellula, vengono buttate fuori attraverso questa P-glicoproteina che non è altro che una pompa di efflusso di determinate molecole. E questo è molto importante perché chiaramente costituisce un meccanismo di protezione filogenetica della cellula staminale, ma è molto importante anche perché giustifica il motivo per cui alcune neoplasie, che originano proprio dalle cellule staminali, non sono sensibili ai farmaci antiblastici o per lo meno non lo sono le cellule staminali stesse, mentre lo sono le cellule più mature che derivano dalla stessa progenie. Ad esempio la staminale leucemica non è sensibile alla chemioterapia e non è sensibile perché, appunto, ha molto ben sviluppato questa P-glicoproteina, che è una pompa che serve a mandare fuori dalla cellula molti farmaci antineoplastici. Man mano che la cellula si differenzia perde questa pompa di efflusso e acquisisce altri antigeni, in particolare il CD38, il CD33 (soprattutto la linea mieloide) e poi tutti i marcatori di linea: a secondo se è una linea linfoide o se è una linea megacariocitica o se è una linea eritroide avrà i diversi marcatori specifici per quella linea. Tuttavia l’aspetto che dobbiamo ricordare è soprattutto il fatto che la cellula staminale emopoietica è positiva per il CD34. Sappiamo che esiste tanta letteratura sulle cellule staminali, per cui di queste cellule staminali si è documentata la potenzialità e la capacità (di quelle che stanno nel midollo) di differenziarsi non soltanto in cellule staminali emopoietiche ma, con opportuni mezzi di coltura e opportuna stimolazione, queste cellule sono in grado anche di differenziarsi in altri tessuti (per esempio il tessuto nervoso, muscolare) e quindi la cellula staminale pluripotente viene considerata una sorte di "fonte di tessuti" (muscolare, epatico, nervoso, ecc) proprio per questa plasticità del midollo osseo, ovvero queste cellule in grado di tornare indietro nel processo differenziativo e differenziarsi verso altri tessuti. Tutto questo ha fatto si che esiste tutta una letteratura di impiego delle cellule staminali dell'adulto, in contrapposizione alle cellule staminali embrionali (qui intervengono aspetti etici e religiosi e quindi si cerca sempre di più di utilizzare le cellule staminali dell'adulto). Le cellule staminali dell'adulto hanno sicuramente dei vantaggi, soprattutto il vantaggio di una disponibilità più ampia o, per lo meno, meno problematica delle cellule staminali embrionali. Diversi studi, anche in altre branche della medicina, stanno cercando di mettere a punto delle metodiche per permettere di espandere queste cellule staminali, di farle differenziare verso dei tessuti specifici e di utilizzarle come una sorta di tessuto di riparazione per determinati danni. * Le ricerche più famose sono quelle fatte sul differenziamento di queste cellule staminali verso i cardiomiociti, condizione questa che permette, appunto, di sviluppare in vivo dei cardiomiociti e che è stato utilizzato per riparare i danni di un'area infartuata. Per cui quando c'è un'area infartuata, dove c'è quindi fibrosi, con l'iniezione di cellule staminali si ripara questo tessuto e quindi queste cellule diventano cardiomiociti. Questo purtroppo succede negli animali di laboratorio, ma non succede nell'uomo, non sappiamo bene per quale motivo. È possibile che nel futuro questi studi possono trovare la chiave di volta della soluzione e utilizzare le cellule staminali come un tessuto capace di differenziarsi verso un altro tessuto ed essere utilizzato per la medicina rigenerativa, ma al momento restiamo con i piedi per terra e vediamo a cosa servono le cellule staminali emopoietiche. Ricordiamo che le cellule staminali stanno nel midollo osseo, posso passare in piccole quantità nel sangue periferico e danno origine a tutta una serie di elementi differenziati che configurano, in questo caso (si riferisce alla slide), la linea mieloide propriamente detta, la quale si conclude con il granulocita neutrofilo che è presente nel sangue in un pool circolante e in un pool marginale e che poi, soprattutto in seguito a stimoli infiammatori (per chemiotassi) va a finire in altri tessuti e serve, appunto, a combattere l'infiammazione. La riserva dei granulociti neutrofili non è soltanto nel sangue periferico, dove abbiamo detto esiste un pool circolante e un pool marginale, ma è presente anche nel midollo dove buona parte del compartimento mieloide propriamente detto è rappresentato da questa riserva di granulociti midollari. Abbiamo visto le condizioni fisiologiche; vediamo le condizioni patologiche. LEUCOPENIA La leucopenia è una riduzione del numero dei globuli bianchi. La leucopenia può essere fondamentalmente dovuta a tre motivi: 1) o aumenta il pool di riserva e marginale. E questa è una condizione abbastanza rara: si vede a volte nei pazienti che hanno una grossa milza, dove c’è una condizione che viene definita ipersplenismo, condizione che determina una sorta di sequestro dei globuli bianchi a livello della milza; 2) condizione molto più frequente è la leucopenia iporigenerativa; 3) l'altra condizione molto frequente è la leucopenia da ridotta sopravvivenza. Cioè, o si producono meno globuli bianchi o se ne distruggono di più. La leucopenia iporigenerativa può essere indotta da: - farmaci, l'esempio classico è quello della chemioterapia che induce una distruzione delle cellule almeno del compartimento maturativo midollare e questo induce quindi una leucopenia iporigenerativa. Ma ci sono anche altri farmaci che su base idiosincrasica inducono questa leucopenia, tipico esempio è il cloramfenicolo, antibiotico che veniva utilizzato quando il professore era bambino e che poi è risultato tossico; - anche l'aplasia o l'ipoplasia midollare, che possono essere congenite o acquisite; - o una invasione midollare da parte di neoplasie oppure da parte di tessuto fibrotico: tutto questo chiaramente toglie spazio alla normale mielopoiesi e quindi determinano leucopenia; - oppure un'ematopoiesi inefficace, per l'incapacità di questo albero maturativo di portare a compimento la maturazione e di fare dei frutti maturi: questa la chiamiamo ematopoiesi inefficace, cioè un difetto della capacità maturativa. Oppure possono esserci condizioni di ridotta sopravvivenza: - si verifica, per esempio, quando si ha una sepsi, perché quando vi è una sepsi si ha un consumo di questi globuli bianchi dovuto al fatto che questi globuli bianchi vanno a finire nei vari tessuti che sono sede di infezione e a volte, per diversi motivi, il midollo non riesce a compensare questo eccesso di perdita; - oppure un'aumentata distruzione nei casi di ipersplenismo che, oltre un sequestro, provoca anche un'eccessiva distruzione dei globuli bianchi; - ma ci possono essere anche dei farmaci che ne facilitano la distruzione con meccanismo immunologico, per cui appunto la distruzione da autoanticorpi contro i globuli bianchi. Esiste anche una pseudoleucopenia: quando diminuisce il pool funzionale e aumenta il pool di riserva, ma anche questa forma è abbastanza rara. Esistono diversi gradi di leucopenia che sono variamente definiti, ma fondamentalmente definiamo una neutropenia grave, lieve e moderata: - Per neutropenia grave si intende un numero di neutrofili al di sotto di 500 per mm3 e questo perché è dimostrato che al di sotto di questa soglia c'è un rischio di infezione molto più elevato. Non solo è possibile la neutropenia, ma è anche possibile la riduzione di eosinofili, che è una condizione abbastanza rara e anche difficile da valutare; una riduzione dei basofili, anche questa rara e di solito passa inosservata. Queste due situazioni sono difficili da osservare perché gli eosinofili e i basofili, in genere, sono abbastanza pochi nella formula leucocitaria e quindi una loro diminuzione viene spesso non visualizzata. Ma, appunto, la cosa importante è la neutropenia, cioè la riduzione dei neutrofili. Riduzione dei neutrofili che, come dicevamo, quando è di grado severo espone a un rischio elevato di infezione, e queste infezioni sono pure infezioni subdole, perché molto spesso il paziente, pur affetto da una grave infezione, non ha una evidenziazione classica dell'infiammazione: la febbre magari è presente, ma vengono a mancare molto spesso il dolor, il rubor e il calor. La febbre, però, è quasi sempre presente e la febbre è un sintomo importante, in sua presenza bisogna subito intervenire. Quindi il paziente presenterà soltanto uno stato di prostrazione, intensi brividi e febbre elevata. In un paziente neutropenico, noi dobbiamo intanto definire qual è il rischio e il rischio varia a secondo del numero dei dei neutrofili. Come dicevamo prima, il rischio è elevato per valori inferiori a 500: questo costituisce il vero rischio infettivo. Il rischio in pazienti che hanno neutrofili fra 500 e 1000 è sicuramente un rischio moderato. Ma non è importante solo la profondità della leucopenia, quello che conta è anche la durata, per cui se un paziente ha una neutropenia che dura 20 giorni chiaramente avrà un rischio molto più alto di un paziente che ha una neutropenia di 10 giorni. Quindi il rischio di infezione è in funzione della gravità e della durata della neutropenia. Ricordiamoci che la febbre potrebbe essere il solo sintomo di questa infezione, perché i sintomi dell'infiammazione sono legati alla presenza dei neutrofili che liberano le sostanze che poi danno luogo ai segni della flogosi, quindi se un paziente è fortemente neutropenico può non avere nessun segno di flogosi e avere soltanto la febbre. In questo caso la terapia antibiotica deve essere fatta d'urgenza e deve essere fatta senza conoscere l'agente eziologico, cioè si deve fare una terapia antibiotica empirica con antibiotico a più ampio spettro possibile. Nel frattempo si mettono in atto tutte le indagini diagnostiche per cercare l'agente eziologico, ma ricordiamoci che non bisogna aspettare di fare la diagnosi eziologica per iniziare la terapia. Il paziente neutropenico va trattato subito, nel giro di poche ore, più tardi si tratta più il paziente rischia la sepsi grave e quindi la morte. · Quali sono le condizioni che provocano una neutropenia? Abbiamo visto che possono essere i farmaci, possono essere localizzazioni di tumori, possono essere infezioni per esempio che provocano un difetto midollare, anche difetti nutrizionali come nei soggetti che hanno carenza di vitamina B12 o di folati. Tutte queste condizioni possono provocare leucopenia. Ovviamente le leucemie si comportano come se fossero delle neoplasie solide, lo stesso la mammella e la prostata: tutte condizioni che determinano un'invasione midollare e quindi impediscono la normale produzione. Tra i farmaci abbiamo già detto come i chemioterapici e i citostatici sono emblematici nell'indurre leucopenia, ma anche altri farmaci possono indurre leucopenia. Quindi quali sono i farmaci in grado di indurre leucopenia? Sono, appunto, quei farmaci in grado di indurre questa agranulocitosi (assenza di produzione di granulociti) e tra questi ci sono gli analgesici antipiretici (antiinfiammatori), il più famoso di questi è la noramidopirina (la famosa novalgina); anche alcuni antistaminici; alcuni diuretici, in particolare la clorotiazide; alcuni farmaci utilizzati nel trattamento delle tireopatie, in particolare il carbimazolo; e alcuni farmaci utilizzati in neurologia come tranquillanti come la carbamazepina. Questi farmaci non li dobbiamo sapere per forza, ma ci servono a capire come è ampio il ventaglio di possibilità di avere una agranulocitosi a causa dei farmaci ed è difficile, quando viene un paziente, riuscire a capire quale farmaco è in grado di indurre questo tipo di effetto collaterale, soprattutto nel paziente che spesso prende tanti farmaci. Questi farmaci sono per altro accomunati dal fatto di contenere un anello benzenico, che è una delle sostanze più tossiche che esistano. Altre cause di neutropenia: - molte infezioni virali possono provocare neutropenia: per esempio il virus di Epstein-Barr, il Citomegalovirus, l'HIV (è molto frequente una presentazione con neutropenia nei pazienti con HIV). Comunque tutti i virus possono dare neutropenia che magari, nella maggior parte dei casi, non è una leucopenia grave, perché magari il paziente ha una buona riserva midollare e ha un buon emocromo per cui l'abbassamento di questi neutrofili non va al di sotto della soglia di affezione. Molto importante è il Parvovirus, perché è un virus decisamente tossico; - alcune infezioni batteriche possono provocare leucopenia. Il paradigma sta nel fatto che le infezioni batteriche provocano spesso leucocitosi neutrofila, questo è quello che succede nella stragrande maggioranza dei casi, perché un'infezione batterica è accompagnata da un aumento dei neutrofili; però ricordiamoci che ci sono alcune infezioni batteriche che possono essere caratterizzate da neutropenia, in particolare la tubercolosi, la brucellosi e il tifo. Quando facciamo la chemioterapia impediamo la formazione degli elementi mieloidi e quindi quello che troviamo nel sangue periferico è la leucopenia. Ma impediamo anche la proliferazione degli elementi mieloidi più maturi e quindi la conseguenza è sempre la leucopenia. In effetti, andando a vedere l'albero di maturazione e di proliferazione delle cellule staminali che si differenziano nelle varie progenie, se noi facciamo la chemioterapia è come se mettessimo una sorta di bomba e distruggiamo prevalentemente la serie mieloide. Ma anche la serie linfoide viene distrutta dalla chemioterapia. Quindi si capisce bene come viene scompaginato il normale sistema di proliferazione delle cellule midollari staminali. L'altro versante è, invece, la leucocitosi. LEUCOCITOSI Per leucocitosi si intende un aumento dei globuli bianchi superiore a 11000 per mm3 con una neutrofilia superiori a 7500 per mm3. Questo può essere dovuto a tante condizioni: - alcuni farmaci. Abbiamo visto farmaci che provocano leucopenia; altri farmaci provocano leucocitosi. In particolare, la condizione più frequente è quella dei cortisonici: non è raro, anzi vengono molto spesso pazienti in ematologia con una diagnosi di leucocitosi e si scopre che questo paziente fa uso di cortisone. Quindi ricordate che il cortisone provoca una leucocitosi neutrofila, perché il cortisone demarginalizza i neutrofili: non è che ne fa produrre di più, ma semplicemente quel pool marginato di neutrofili diventa un pool circolante. Anche l'adrenalina agisce con lo stesso meccanismo; - altre condizioni come lo stress, l'attività fisica: tutto questo provoca una demarginazione di quel pool marginato, ma in questo caso l'aumento di neutrofili è abbastanza modesto; - come dicevamo prima, le infezioni batteriche acute, nella stragrande maggioranza dei casi, provocano leucocitosi neutrofila; - quando ci sono dei danni ai tessuti o quando c'è un'infiammazione chiaramente si ha un aumento della leucocitosi neutrofila; - chiaramente anche la splenectomia, perché la milza è la sede dove avviene la distruzione dei globuli bianchi e togliendo la milza si toglie una sede importante di distruzione. Quindi è logico che aumenta la presenza dei globuli bianchi. Nella progressione della maturazione della cellula staminale fino a granulocita neutrofilo, se il midollo si mette a produrre più elementi mieloidi, molti elementi mieloidi immaturi passano anche nel sangue periferico e, nel sangue periferico, osserviamo la presenza di neutrofili un pochino meno maturi: allora, si dice che nel paziente c'è uno shift maturativo a sinistra, un'espressione per dire che sono presenti più neutrofili immaturi in un paziente che produce, appunto, una maggiore quantità di neutrofili. Però queste sono delle definizioni ormai abbastanza abbandonate. Viceversa, quando i neutrofili sono particolarmente maturi, come succede per esempio in casi di carenza di vitamina B12, si parla di shift maturativo a destra. Esistono anche dei difetti qualitativi dei globuli bianchi, oltre che quantitativi. I difetti qualitativi, nella maggior parte dei casi, sono congeniti, ma possono essere anche acquisiti: - per esempio, come abbiamo accennato prima, nella carenza di vitamina B12 osserviamo questa ipersegmentazione dei neutrofili (eccessiva maturazione) e questo è un difetto qualitativo; - oppure nelle infezioni osserviamo un aumento dei granuli e queste sono delle granulazioni tossiche che si osservano, appunto, nei neutrofili nel corso di infezioni; - oppure c'è la cosiddetta anomalia di Pelger-Huet, che usualmente è una condizione congenita, ma ci possono essere anche delle anomalie acquisite, ma che comunque non hanno un significato funzionale ma morfologico: i neutrofili hanno tipico aspetto a occhiale, sono presenti due lobi nucleari; - ci possono essere dei deficit di mieloperossidasi e in questo caso sono condizioni di solito congenite. Come sappiamo la mieloperossidasi è presente all'interno dei neutrofili ed è una potente arma antibatterica; - l'anomalia di May-Hegglin, che è un altro difetto autosomico dominante che, oltre a caratterizzarsi per la presenza di inclusioni citoplasmatiche, si caratterizza per il fatto che questi pazienti hanno delle piastrine giganti. Parliamo adesso dei difetti appartenenti alla linea linfoide. LINFOCITOPENIA Si definisce linfocitopenia una riduzione dei linfociti sotto i 1500 per mm3 negli adulti e meno di 3000 per mm3 nei bambini. Ricordiamoci che quando parliamo di linfocitopenia, leucopenia, ecc... non ci riferiamo mai alla valutazione percentuale, ma ci riferiamo sempre a una valutazione del valore assoluto. Bisogna fare attenzione a ciò, perché molto spesso gli emocromi vengono dati con una formula in percentuale che può essere fuorviante. Quello che dobbiamo andare a guardare sono sempre i valori assoluti, perché nella formula percentuale la somma deve fare sempre 100: quindi è chiaro che se, per esempio, si abbassano i neutrofili aumentano percentualmente i linfociti. Spesso ci sono pazienti che arrivano con una diagnosi di linfocitosi, ma che hanno, invece, una neutropenia, perché se ho 30% di neutrofili la formula leucocitaria che deve per forza fare 100 fa aumentare i linfociti e allora avremo il 60% di linfociti. Pertanto ricordiamo che bisogna considerare non le percentuali ma il valore assoluto!! Tornando al discorso della linfocitopenia, cosa può determinare linfocitopenia? - terapia con steroidi: abbiamo detto che gli steroidi fanno aumentare i neutrofili, perché li demarginalizzano, ma contemporaneamente fanno ridurre i linfociti, perché lo steroide è un potente linfocitolitico; - alcune infezioni virali o batteriche possono provocare una linfocitopenia, l'esempio più classico per le infezioni virali è l'HIV; - alcune neoplasie, per esempio il linfoma di Hodgkin. Il linfoma di Hodgkin è una neoplasia linfoproliferativa e spesso agli esami si dice che il quadro periferico nel linfoma di Hodgkin presenta un aumento dei linfociti. No! È esattamente il contrario, c'è una riduzione dei linfociti, perché probabilmente questi linfociti vanno a finire tutti quanti nel linfonodo sede di malattia. In effetti non si sa bene quale sia il meccanismo, ma sicuramente i pazienti con linfoma di Hodgkin hanno la linfocitopenia; - ovviamente se ci sono altre malattie che infiltrano il midollo; - alcune malattie croniche possono causare linfocitopenia. LINFOCITOSI La linfocitosi è invece una condizione abbastanza frequente e anche questa può essere assoluta o relativa: però noi dobbiamo sempre basarci sul numero assoluto; la linfocitosi relativa è sempre secondaria ad altre condizioni: per esempio la neutropenia fa abbassare la percentuale di neutrofili e fa aumentare i linfociti, ma è un aumento relativo. Quello che a noi interessa è la linfocitosi assoluta. La linfocitosi assoluta che si può venire a creare per tutta una serie di malattie virali, per esempio la parotite, l'influenza, la varicella, l'herpes; qualche malattia non virale, tipo la brucellosi. Ma diciamo che la tipica malattia che provoca linfocitosi è la mononucleosi infettiva o le sindromi mononucleosiche, dove ci si attende un Citomegalovirus o altri virus. E chiaramente, l'altra linfocitosi assoluta è legata a tutte le malattie linfoproliferative che hanno una presentazione nel sangue periferico: il prototipo di queste malattie è la leucemia linfoide cronica. Lezione 3 ANEMIE La scorsa volta abbiamo parlato dell'eritropoiesi. Se ricordate l'eritropoiesi è caratterizzata da una elevatissima produzione di globuli rossi, fino a 3 milioni al giorno per chilogrammo e tutto dipende dalla cellula staminale pluripotente che da origine a cellule più commissionate (committed) che prendono stretti rapporti con lo stroma midollare (cellule adipose, fibroblasti, osteoblasti, osteoclasti ed altre) e tutto questo processo viene regolato dall' eritropoietina, ormone prodotto dal rene che agisce non tanto sulla cellula staminale o sugli elementi più maturi della serie eritroide ma sulle BFU-E e CFU-E, questi sono i bersagli preminenti dell' eritropoietina. Tale ormone determina una notevole amplificazione di tutto l'eritrone fino alla produzione degli eritrociti, preceduti nelle tappe maturative dai reticolociti. Tutto questo ci serve ad introdurre il concetto di anemia. L'anemia è una condizione che incontrerete molto spesso qualunque mestiere facciate, anzi potremmo dire che molte condizioni anemiche riguardano altri specialisti piuttosto che l'ematologo. L'approccio ad un paziente anemico è estremamente semplice, almeno nelle prime fasi della diagnostica, anche se molti medici non ne sono capaci. Quando parliamo di anemia non consideriamo mai il numero dei globuli rossi ma il valore dell'emoglobina: il paziente non è anemico perché ha un numero di globuli rossi basso ma perché è diminuita la concentrazione di emoglobina al di sotto del range fisiologico. Per convenzione si parla di anemia quando la concentrazione di emoglobina è inferiore a 14g/dL nell'uomo ed a 12g/dL nella donna, ovviamente queste sono definizioni rigide che devono essere sempre vagliate rispetto alla situazione contingente del paziente. E' chiaro che se una donna aveva 16 g di emoglobina e passa a 12g, possiamo già parlare di anemia anche se il dato richiesto come criterio diagnostico non è completamente soddisfatto. Quindi ricordate sempre di non considerare i valori in maniera assoluta ma di modularli sulla situazione del vostro paziente. Fondamentalmente esistono tre possibilità che possono condurre ad un quadro anemico. 1. Una è la perdita di sangue, qualsiasi condizione emorragica può determinare anemia. 2. La seconda causa è un eccesso di distruzione dei globuli rossi 3. la terza è rappresentata dalla ridotta produzione dei globuli rossi. Quindi quando approcciate un paziente affetto da una condizione di anemia dovete sempre collocare il paziente in una di queste tre condizioni. E' chiaro che una volta iniziato il percorso diagnostico, dopo aver collocato il paziente in una delle tre possibili cause, dovete continuare ad indagare. Così l'emorragia (quando c’è una perdita di sangue) potrà essere causata da una perdita acuta o cronica di sangue; oppure l'emolisi (aumentata distruzione) che può essere causata dall'anomala morfologia del globulo rosso (anomalie intrinseche del globulo) o da altra causa che agisce all'esterno del globulo stesso. Quando c'è una ridotta produzione, questa potrà essere dovuta a ridotta proliferazione della cellula staminale oppure a mancata proliferazione della serie eritroide (eritroblasti) e tutta una serie di altre condizioni che poi vedremo. Quindi da ricordare l'importanza nella prima fase del percorso diagnostico di collocare il paziente in una di queste tre possibili cause. Già con il semplice esame rappresentato dalla conta dei reticolociti si può discernere una condizione dalle altre due (vedremo meglio in seguito). Quindi nella diagnostica di tutte le condizioni di anemia è importante valutare la conta dei reticolo citi. Tuttavia oltre alla conta dei reticolociti è importante anche andare a definire le anemie sulla base del volume globulare medio cioè di quanto è grande questo globulo. Il volume globulare medio ci permette di classificare le anemie in: microcitiche, normocitiche e macrocitiche. Valutando questo parametro, che tutti i contaglobuli ormai ci danno, già questo serve ad indirizzarci nella diagnosi. - Ad esempio le anemie microcitiche, in cui il globulo rosso ha un volume inferiore ad 80 fL, fondamentalmente sono dovute a carenza di ferro o alla talassemia (quest’ultima infatti viene anche detta microcitemia). Tra queste due condizioni sarà importante fare una diagnosi differenziale e poi vedremo come. - Le anemie macrocitiche in cui il globulo rosso ha un volume globulare medio superiore a 100 fL, anche in questo caso le possibilità diagnostiche sono poche perché fondamentalmente un’anemia macrocitica può essere da carenza di acido folico o vitamina B12 oppure può essere conseguente a mielodisplasia o da malattia epatica. Quindi il volume globulare medio rappresenta un parametro semplice da valutare che ci indirizza nella diagnosi in maniera specifica e la rende anche agevole. Quando il volume globulare medio è intermedio e cioè tra 80 e 100 fL l'algoritmo diagnostico suggerisce di andare a valutare il parametro che dicevamo prima e cioè la conta dei reticolo citi: - se i reticolociti sono aumentati non potrà che essere un'anemia emolitica o emorragica. - se i reticolociti sono bassi o normali bisognerà andare a studiare il midollo per vedere se c'è qualche problema al midollo. Quindi la conta dei reticolociti insieme al volume globulare medio diventa un elemento cruciale nella valutazione del paziente. Gli esami di laboratorio che bisogna fare nella diagnostica di routine di anemia sono in numero relativamente ristretto perché già l'esame emocromocitometrico normale ci fornisce i valori di: emoglobina, ematocrito, indice reticolocitario, MCV (volume globulare medio), conta e formula leucocitario ed il numero di piastrine. Bisognerà inoltre valutare la morfologia degli eritrociti perché sarà utile nella diagnosi. Vi sottolineo nuovamente l'importanza della conta reticolocitaria e valutare inoltre sideremia ed i valori di ferritina e transferrina con la TIBC (total iron binding capacity, cioè la capacità di legare il ferro propria della transferrina deputata al trasporto di ferro). NORMALE CARENZA MARZIALE ANEMIA SIDEROPENICA EMOCROMATOSI E EMOSIDEROSI SIDEREMIA 80-150 < 80 < 30 > 150 TRANSFERRINEMIA 200-300 > 300 > 400 < 200 SATURAZIONE TRANSFERRINA 20-40% < 20% < 10% > 75% FERRITINEMIA 50-200 < 20 < 10 > 1000 Se un paziente avrà una carenza di ferro avrà sideremia e ferritina bassa e una elevata TIBC espressione di un'aumentata avidità nel legame del ferro e questo aspetto come visto è importante soprattutto nelle anemie microcitiche (proprio quelle che caratterizzano l’anemia sideropenia). Sono ancora importanti i valori di bilirubina e LDH. La bilirubina infatti rappresenta il prodotto catabolico della distruzione degli eritrociti e i livelli di bilirubina (indiretta) aumenteranno nei pazienti con anemia dovuta ad eccessiva distruzione eritrocitaria e aumenterà anche l’LDH come succede in tutte le patologie in cui c’è una distruzione cellulare. Se noi facciamo questi esami abbiamo un indirizzo diagnostico preciso sulla condizione di anemia: naturalmente questi sono esami di primo livello, è chiaro che se poi noi documentiamo bilirubina indiretta ed LDH aumentati sarete in una condizione di anemia emolitica però dovrete poi indagare sulle condizioni che hanno determinato l'emolisi. · Un altro parametro che valutiamo oltre l'MCV è l'RDV o indice di anisocitosi (differenza di volume) che ci serve nella diagnostica differenziale tra sideropenia e talassemia. Conta dei reticolociti E’ importante soffermare la nostra attenzione sull’importanza della conta dei reticolociti. I reticolociti sono caratterizzati da un reticolo citoplasmatico rappresentato da sostanza granulofilamentosa dovuta a residui di RNA , hanno densità inferiore rispetto ai globuli rossi ed esprimono il recettore per la transferrina che è invece assente nei globuli rossi. Ancora oggi molto spesso i laboratori analisi valutano la conta reticolocitaria soltanto in percentuale considerando normali i valori compresi tra 1 e 2%. Noi non ci dobbiamo però fermare al valore percentuale ma dobbiamo chiedere il valore assoluto oppure lo dobbiamo calcolare noi stessi facendoci i conti. Una valutazione in percentuale può essere infatti fuorviante!! Infatti se otteniamo il 3% di un numero normale di globuli rossi (5 milioni) cioè 150000 reticolociti che sicuramente rappresenta un numero aumentato di reticolociti perché normalmente i reticolociti vanno da 50000 a 100000 unità. Se invece io ho il 3% di 2 milioni di globuli rossi, ovvero 60000 reticolociti, ho un valore normale di reticolociti mentre il dato in percentuale mi potrebbe ingannare facendomi credere che la conta reticolocitaria sia aumentata. Se questo stesso paziente con 2 milioni di globuli rossi ha l'1% di reticolociti il laboratorio vi darà questo dato come normale quando invece sarà un valore più basso della norma. Quindi quando valutate la conta reticolocitaria fate attenzione che i dati che valutate siano espressi in valore assoluto e non percentuale o se il laboratorio vi dovesse fornire il dato in percentuale fatevi i conti e risalite al dato assoluto. Si può anche fare il cosiddetto valore corretto dei reticolociti perché in realtà i reticolociti stanno a cavallo fra midollo e sangue periferico ed è calcolato che stiano un giorno nel sangue periferico prima di diventare globuli rossi. E' chiaro che se si verifica un'aumentata dismissione di reticolociti un po’ più immaturi nel sangue periferico per via del fatto che il midollo risponde allo stimolo anemico, questi reticolociti staranno nel sangue più di un giorno (due o tre giorni) e quindi c’è anche una formula che calcola l'indice reticolocitario tenend conto del fatto che si tratta di reticolociti più immaturi e quindi persistono più a lungo nel sangue periferico. Questa è comunque una complicazione da specialisti invece è importante tenere a mente la differenza tra valore assoluto e percentuale di reticolociti. Non tutte le anemie sono uguali. Esistono anemie lievi, moderate e gravi. In alcune classificazioni ad esempio un valore nell’uomo di 13 g di Hb non è considerata condizione anemica, questo perché è difficile dare dei cut off molto precisi, in generale però: - consideriamo anemia lieve fino a 10 g di Hb - fino a 8 g un’anemia moderata - < 8 g un’anemia grave Otto grammi è anche il limite al di sotto del quale è indicata la trasfusione di sangue. Anche in questo caso comunque non si tratta di un dogma, il prof vede molto spesso pazienti che si rivolgono a lui, mandati dal medico curante, con meno di otto grammi di emoglobina per fare una trasfusione. E' vero che 8g di emoglobina rappresenta il cut-off per fare una trasfusione, ma se il paziente ad esempio è in una condizione di anemia sideropenica basta iniziare la terapia con sali ferrosi perché il paziente migliori senza il ricorso alla trasfusione; lo stesso discorso vale per la carenza di folato o vitamina B12. Al contrario può esservi un paziente cardiopatico o cerebropatico che ha bisogno di una maggior quota di globuli rossi nel sangue e che quindi potrà ricevere una trasfusione anche se i valori di emoglobina sono superiori ad otto grammi. Quindi il valore di otto grammi/dL è indicativo per eseguire una trasfusione ma a 7,9 grammi non scatta la mezzanotte di Cenerentola per fare la trasfusione!!Ricordate sempre che si tratta di parametri indicativi. SINTOMI E SEGNI Lo stato anemico può essere caratterizzato da tanti sintomi e segni. Il primo segno è il pallore. Tenete conto però che il colore della pelle non dipende soltanto dai globuli rossi ma anche da altri fattori, ad esempio un paziente iperteso avrà un colorito più rosso. Alcuni pazienti hanno un colorito bluastro per via di cardiopatie o insufficienza respiratoria. Ci sono pazienti che hanno un colorito giallastro perché sono di razza asiatica o perché hanno l'ittero che compare nelle anemie emolitiche ed è a prevalente bilirubina indiretta. Alcuni sintomi possono fornire degli indizi importanti: → Il colorito giallo-limone della cute è abbastanza specifico di anemia megaloblastica da carenza di B12. → Le ulcere alle gambe sono abbastanza frequenti e specifiche nella drepanocitosi o anemia falciforme. → Le ragadi labiali, le unghia fragili fino alle unghie a cucchiaio (coilonichia) sono tipiche delle anemie ferroprive. → La glossite con disepitelializzazione della lingua la ritroviamo nei deficit di B12 e nelle anemie sideropeniche. Il pallore cutaneo può essere visto con più facilità nelle pliche della mano che risalteranno di più perché già la mano è un pò più chiara da sé. Una attenta valutazione di cute e mucose è molto importante nella diagnosi di anemia. → La trombosi si può ritrovare nella drepanocitosi e nell'emoglobinuria parossistica notturna. Il paziente con anemia può essere tachicardico ed avere dispnea da sforzo ed avere episodi di angina pectoris o abdominis e la claudicatio. Anche nervosismo ,irritabilità , cefalea e vertigini, acufeni e scotomi, molti soggetti che soffrono di anemia sideropenica presentano cefalea. E' chiaro che se il sangue scorre con una viscosità diminuita ci possono essere dei soffi cardiaci, il famoso soffio anemico, questo perché nel soggetto anemico il sangue ha una differente fluidità rispetto al sangue normale, la viscosità è minore. L'aumento cronico della gittata cardiaca nei pazienti cronicamente anemici può portare ad ipertrofia ventricolare e condurre fino allo scompenso cardiaco. Si può inoltre avere debolezza, astenia, e può comparire anche una febbricola nei pazienti che sono gravemente anemici. La dispnea nelle anemie più gravi, con valori di emoglobina inferiore a tre grammi, può anche insorgere a riposo e si può giungere nelle fasi terminali a collasso e coma, di può morire di shock anemico! → Se un paziente si presenta con un quadro di alterazione del sistema nervoso centrale e/o soprattutto periferico dovete subito pensare al deficit di vitamina B12, poiché questa vitamina non serve soltanto a fare globuli rossi ma serve anche al sistema nervoso centrale e periferico. Alterazioni a livello gastroenterico ( nausea, stipsi, diarrea, dispepsia) sono frequenti nella carenza di vitamina B12, anzi ne sono la causa! La disfagia insorge nel deficit di B12 e nella carenza di ferro. Se il paziente oltre ad essere anemico ha anche una splenomegalia dobbiamo pensare ad una condizione di emolisi, talassemia o ad una neoplasia della milza che causa anche anemia. In caso di anemia falciforme può comparire dolore addominale, al torace, agli arti inferiori. Tutti questi aspetti possono avere una differente gravità , cioè la gravità dell'anemia non dipende soltanto dai valori dell'emoglobina. Quando siamo di fronte ad un paziente anemico dobbiamo stabilire la gravità della situazione che dipende da tante cose: bisogna prima di tutto escludere che ci siano delle false anemie o cosidette anemie spurie, poiché in tutte quelle condizioni in cui si ha un aumento della parte liquida del sangue rispetto alla parte corpuscolata, c'è una condizione di anemia. A parte lo scompenso congestizio che è una condizione abbastanza rara, la gravidanza rapperesenta una condizione di pseudoanemia perché le donne in gravidanza hanno un aumento della parte liquida del sangue con diminuizione relativa della parte corpus colata, quindi c’è una condizione fisiologicamente di anemia spuria. Anche l'eccessiva idratazione può portare ad un quadro di anemia spuria per sbilanciamento delle due componenti ematiche ovvero quella corpuscolata e quella liquida. E' molto importante inoltre nell'anamnesi escludere perdite di sangue evidenti (ematemesi, melena, traumi recenti, emottisi, epistassi, perdite ginecologiche o genitourinarie molto spesso causa di anemia sideropenica, prelievi ripetuti). A noi è capitato molto spesso di avere pazienti con turbe psiachitriatiche che si autosalassavano e che come conseguenza sviluppavano anemia senza che riuscissimo a capirne la causa. E' molto importante la raccolta anamnestica anche geografica di provenienza, perché dalle nostre parti è endemica la talassemia. In un paziente con anemia microcitica la prima cosa da chiedere è se in famiglia si ha familiarità per talassemia. Chiedete inoltre al paziente se ha avuto episodi di ittero o subittero, perché le anemie emolitiche possono manifestarsi con questo colorito giallastro della cute. L'altra volta è stato detto di quanto sia devastante la chemioterapia per l'effetto tossico sull'intero midollo e quindi anche verso i precusrsori eritroidi. Un paziente che fa chemioterapia diventa anemico, purtroppo è la regola. Ci sono farmaci che possono indurre anemia ed anche alcune malattie sistemiche. · La cosa più importante nel valutare l'anemia è la rapidità di insorgenza del quadro anemico. I pazienti con un’anemia che insorge molto rapidamente sono pazienti che stanno male anche per valori di emoglobina abbastanza alti, invece pazienti in cui l’anemia insorge molto lentamente possono anche non avere alcun sintomo!! Per cui non è inverosimile trovarsi di fronte a pazienti con 6 grammi di emoglobina e che non avvertono una sintomatologia peculiare, anche se poi indagando più approfonditamente vi accorgete che hanno la dispnea appena fanno due gradini tuttavia magari in condizioni di riposo non avvertono alcun sintomo. * Il prof racconta sempre l’aneddoto di una vecchietta con 3 grammi di emoglobina ed alla quale chiesi come stava e lei mi rispose :"Sono il ritratto della salute". La vecchietta veniva servita da tutto un corteo di figli, nipoti e pronipoti e quindi non facendo nessuno sforzo non accusava alcuna sintomatologia, proprio perché l’anemia era insorta molto lentamente. Se l'anemia insorge lentamente l'organismo mette in moto dei meccanismi di compenso che limitano la sintomatologia. · Altri aspetti molto importanti sono l'età e le condizioni generali e la funzionalità del sistema cardio-cerebro-vascolare: un paziente cardiopatico ha bisogno di un maggiore valore di emoglobina; se un paziente ha una vascolopatia cerebrale e quindi una ipossia cerebrale cronica, chiaramente ha bisogno di un apporto maggiore di ossigeno al cervello e pertanto risente di più di una condizione di ipossia legata a una carenza di emoglobina. Striscio di sangue periferico Indicazioni importanti possono essere fornite dallo striscio periferico che viene fatto dall’ematologo o raramente dal medico di laboratorio e non dal medico generale. L’altra volta abbiamo visto come l’eritrocita normale ha una forma biconcava ma esistono delle variazioni morfologiche degli eritrociti che sono specifiche di alcune condizioni patologiche. - --- -- L’acantocita che presenta delle caratteristiche spicule, è abbastanza tipico delle condizioni da malassorbimento. - Lo sferocita, una sorta di globulo rosso a palla, chiaramente è presente nella sferocitosi ereditaria ma lo possiamo trovare anche nelle anemie emolitiche autoimmuni in cui i reticolociti immessi nel circolo assumono la morfologia sferocitica. - Ci sono gli schistociti o schizociti che sono estremamente importanti perché non sono altro che frammenti di globuli rossi: osservare questi frammenti ci indirizza verso diagnosi importanti, quadri patologici come la porpora trombotica trombocitopenica (patologia che vedremo meglio in seguito) che è mortale nell’80% dei casi se non fate la diagnosi ma che potete guarire nell’ 80% dei casi se invece riconoscete la patologia. In questa patologia si formano dei piccoli trombi nei vasi più piccoli (esattamente come succede nella CID) ed i globuli rossi al passaggio in questi vasi vengono rotti dai tralci di fibrina del trombo stesso, pertanto quello che vediamo nello striscio di sangue periferico sono i frammenti di questi globuli rossi tagliati passando attraverso le maglie di fibrina → questa è una condizione che ci permette di definire la cosiddetta anemia emolitica microangiopatica in cui la formazione di trombi all’interno dei vasi determina l’emolisi dei globuli rossi. Anche la CID o le vecchie protesi valvolari cardiache appartengono a questa categoria. Quindi trovare gli schistociti nello striscio periferico rappresenta la prova che un trauma ha provocato la frammentazione del globulo rosso. - L’ellissocita è meno importante, esiste l’ellissocitosi ereditaria ma molte altre condizioni possono manifestarsi con l’ellissocitosi. - Molto più importanti invece sono i drepanociti o globuli rossi a falce che si ritrovano nella anemia falciforme o drepanocitosi. - I globuli rossi a bersaglio o codociti si possono riscontrare in diverse condizioni anemiche mentre è abbastanza specifico e quasi patognomonico di mielofibrosi idiopatica (condizione in cui il midollo osseo viene sostituito da tessuto fibrotico) il dacriocita o globulo rosso a lacrima. Quindi potete vedere come l’esame morfologico ha un ruolo importante nella diagnostica delle anemie. Ci vuole un occhio allenato per riconoscere questi aspetti e molti laboratoristi ne sono capaci anche se i moderni analizzatori sono in grado di riconoscere alcuni di questi aspetti. Ritornando alla classificazione fisiopatologica delle anemie come visto è importante definire se il paziente ha: - perdita di sangue - aumentata distruzione - ridotta produzione Per iniziare questo percorso diagnostico i reticolociti sono di estrema importanza, se abbiamo una buona conta di reticolociti escludiamo una condizione di ridotta produzione, pertanto la prima distinzione viene fatta assolutamente attraverso la conta dei reticolociti. La seconda distinzione viene fatta attraverso MCV invece ci fa distinguere le anemie in microcitiche, normocitiche e macrocitiche: · Le anemie microcitiche sono fondamentalmente riconducibili alla carenza di ferro ed alla talassemia. Perché il paziente diventa anemico se manca il ferro? Il ferro è fondamentale nella produzione di emoglobina; il ferro è contenuto nella carne rossa, nei crostacei, nella frutta secca e nei legumi. Il ferro è presente nel fegato dove si deposita ed anche nell’uovo in cui però la presenza di altre sostanze ne impedisce l’assorbimento. Viene perso normalmente attraverso le urine, le feci ed il sanguinamento e ha un basso rate di assorbimento. Viene assorbito nell’intestino, passa nel sangue legandosi alla transferrina e da qui viene trasferito al midollo per fare emoglobina, ai muscoli per fare mioglobina ed a molte cellule perché il ferro entra nella formazione di numerosi enzimi mitocondriali. Ecco perché chi ha una carenza di ferro non ha solamente l’anemia ma ha tutto un corteo sintomatologico che è causato dalla deficienza di ferro negli altri distretti. Il ferro derivante dalla distruzione dei globuli rossi viene riciclato ed i depositi di ferro si accumulano soprattutto nel fegato sottoforma di ferritina. Per produrre globuli rossi c’è pure bisogno di acido folico e vitamina B12 che vedremo meglio in seguito. All’interno del globulo rosso il ferro dovrà localizzarsi in domini idrofobici della molecola di emoglobina perché altrimenti si ossida. Il ferro è contenuto all’interno dell’eme che fornisce un microambiente il quale fa in modo che il ferro non venga idratato ( il ferro arrugginisce e se questo succede nel globulo rosso la situazione diviene incompatibile con la vita!). Si calcola che nell’uomo siano presenti 3,5 grammi di ferro mentre nella donna 2,5. Questo ferro è in parte funzionale (contenuto nell’emoglobina, nella mioglobina e negli enzimi mitocondriali), una grossa quota è il ferro di deposito (identificato come ferritina ed emosiderina, anche se la ferritina rappresenta un ferro di deposito più rapidamente disponibile rispetto all’emosiderina) ed infine il ferro è legato alla molecola di trasporto che è la transferrina ma in piccolissima quota; la transferrina in realtà può legare solo poco ferro per volta. Questi aspetti sono importanti per capire la terapia di ferro che bisogna fare. * La transferrina, proteina prodotta dal fegato, appartiene alle beta-globuline e viene indicata come TIBC (total iron binding capacity) che rappresenta la capacità totale di legare il ferro da parte dell’organismo e dunque l’avidità dell’organismo nel legare il ferro. La transferrina viene inoltre indicata come transferrina totale e transferrina satura. La saturazione normale della transferrina è compresa tra il 20 ed il 45%. Se abbiamo un eccesso di transferrina ed una carenza di ferro naturalmente la saturazione della transferrina diminuirà. E’ importante l’assunzione del ferro in tutte le condizioni in cui viene consumato più ferro come la gravidanza. Se guardate al grafico in cui sono raffigurate le curve di richiesta di ferro in base all’età potete notare che nell’infanzia c’è un picco di richiesta di ferro che in seguito si abbassa un po’ per poi ritornare a crescere gradualmente durante l’adolescenza. Quindi durante questo periodo se il bambino si nutre poco, come avviene nel terzo mondo, o presenta deficit nell’assorbimento (malassorbimento) si può sviluppare un’anemia da carenza di ferro. Non è infrequente che noi ematologi facciamo diagnosi di celiachia in pazienti che si recavano da noi per anemia da carenza di ferro: si tratta di pazienti che non assorbono il ferro e in questi casi quando il paziente non risponde alla somministrazione di ferro per via orale vi si deve accendere subito la lampadina e dovete pensare ad una causa di malassorbimento, una delle più frequenti è la celiachia! Quindi pazienti che non sanno di essere celiaci, perché non hanno una sintomatologia importante, il cui primo sintomo è proprio l’anemia sideropenica. Anche l’adolescente, che spesso cresce molto rapidamente, può presentare carenza di ferro perché spesso l’alimentazione non è adeguata alle richieste dell’organismo in crescita. A questo punto in cui le richieste di ferro tra l’uomo e la donna divergono perché le donne hanno le mestruazioni e con queste perdono molto ferro, se poi la donna va in gravidanza chiaramente il fabbisogno di ferro aumenta molto di più. Alla menopausa le curve nella richiesta di ferro tra uomo e donna ritornano uguali. Questa curva ci fa capire perché l’anemia da carenza di ferro riguarda soprattutto le donne in età fertile perché è qui la maggiore richiesta di ferro. Il ferro entra sia come ferro bivalente che come ferro trivalente, perché così è contenuto negli alimenti, passa l’acidità dello stomaco e viene assorbito a livello del piccolo intestino soprattutto a livello duodenale (diversi organi tra cui pancreas, fegato,ghiandole salivari, stomaco sono importanti per l’assorbimento del ferro). Ci sono alcuni alimenti favorenti l’assorbimento del ferro (la vitamina C) mentre altri come il caffè lo inibiscono. Il ferro viene assorbito dalle cellule intestinali e da qui passa nel sangue → il ferro viene captato dalla transferrina che porta il ferro in tutti quei distretti in cui serve e tra questi il midollo osseo → nel midollo osseo c’è l’eritrone e tutte le cellule immature della serie eritroide fino al reticolocita esprimono recettori per la transferrina che servono ad internalizzare il ferro per utilizzarlo nella sintesi dell’eme (il recettore per la transferrina si mantiene fino al reticolocita, solo il globulo rosso non ha recettori per la transferrina). Tutto ciò viene regolato da una serie di molecole che sono conosciute da poco tempo: la proteina DMT1 serve a fare entrare il ferro bivalente nell’enterocita mentre la ferroportina serve ad immetterlo nel torrente circolatorio. Tutto ciò è regolato dalla epcidina (che regola negativamente la ferroportina ed il passaggio di ferro nel plasma) sintetizzata dal fegato e da altre proteine, tra cui il recettore solubile per la transferrina, che regolano un minore o maggiore assorbimento di ferro. Se c’è una condizione di carenza di ferro allora aumenta la produzione di ferroportina che permette al ferro dei depositi di passare nel plasma ed allo stesso tempo di fare passare più ferro dalla cellula intestinale verso il plasma. Tutto questo serve a dirci che in condizioni di carenza di ferro aumentano le proteine che facilitano l’apporto di ferro al plasma, con riduzione invece della epcidina che come visto svolge un’azione contraria. Il ferro di deposito si accumula prevalentemente nel fegato ed in altri tessuti. · Quali sono le condizioni in cui è richiesto una maggiore quantità di ferro? - Il ferro è maggiormente richiesto nei sanguinamenti, ad es. tutti i disturbi gastroenterici con perdita di sangue, un eccesso di ciclo mestruale in frequenza e/o abbondanza, perdite di sangue dall’albero respiratorio, donazioni eccessive (soprattutto in passato quando il sangue si vendeva), perdite urinarie, pazienti che fanno dialisi, pazienti psichiatrici che si tolgono il sangue da soli, rappresentano situazioni potenzialmente in grado di condurre ad anemia ferropriva. - Un’altra condizione è l’emoglobinuria parossistica notturna in cui l’emoglobina viene persa con le urine (emoglobinuria) e quindi si può sviluppare una carenza di ferro. Anche condizioni fisiologiche come il rapido accrescimento, la gravidanza o l’allattamento, se non c’è un adeguato apporto di ferro, possono portare a carenza di ferro con conseguente anemizzazione. - I pazienti con alterata funzione gastrica (acloridria) o precedentemente gastrectomizzati (trattamento delle ulcere gastriche in passato) possono sviluppare un quadro anemico. In realtà lo stomaco non rappresenta la sede di assorbimento ma è necessaria l’acidità gastrica per ridurre il ferro trivalente a ferro bivalente che poi verrà assorbito. L’acloridria non rappresenta un quadro infrequente al giorno d’oggi perché i pazienti abusano degli inbitori di pompa (omeprazolo, pantoprazolo), come se fossero caramelle → questa è una condizione che provoca acloridria e quindi uno scarso assorbimento del ferro. Quindi una carenza di ferro si può avere anche soltanto per un eccesso di farmaci inibitori di pompa che vengono abusati solo perché essendo esenti dal pagamento del ticket i pazienti li prendono senza alcuna limitazione, tra l’altro i medici lo prescrivono spesso senza motivo. - Ci sono situazioni estremamente rare come la carenza congenita di transferrina o anticorpi contro la transferrina. Se avete un paziente con carenza di ferro a cosa dovete pensare come prima causa? Qual è la condizione più frequente che provoca carenza di ferro? La condizione più frequente, nel 70% dei casi, è una perdita eccessiva di sangue. Nella donna e nell’età fertile la perdita più frequente è quella mestruale e quindi dovete indagare durata, frequenza ed entità del ciclo mestruale della paziente. In un uomo di 50 anni chiederete se ha problemi intestinali perché come seconda causa compaiono le perdite gastroenteriche. Tante neoplasie del colon, soprattutto del colon ascendente, vengono diagnosticate perché il primo sintomo è l’anemia sideropenica. Se infatti la neoplasia cresce nel colon di sinistra, in cui le feci sono già abbastanza formate questa darà subito segno di sé con stipsi o diarrea. Nel colon destro invece la patologia sarà clinicamente silente fino alle fasi avanzate tanto che spesso l’anemia rappresenta il primo sintomo (perché queste neoplasie sanguinano). In questi casi dovete far fare al paziente la ricerca del sangue occulto nelle feci. Anche le emorroidi sanguinanti, l’ulcera peptica, i diverticoli e l’ernia iatale possono portare a quadri di anemia. L’ernia iatale non è altro che lo scivolamento dello stomaco attraverso l’orifizio diaframmatico e quindi lo stomaco risale in cavità toracica. Lo stiramento che questa porzione di stomaco subisce a livello dello orifizio diaframmatico porta a microerosioni con sanguinamento. Se un paziente ha una ectasia dei seni venosi nasali e sanguina un giorno si ed un giorno no, è inutile far fare la coprocultura perché l’anemia è conseguenza delle epistassi ripetute. Alcune situazioni rare sono rappresentate dalla sprue, dal morbo di Chron, dalla carenza di vitamina C ( scorbuto) o dall’eccesso di calcio. * Una volta io ho incontrato un paziente con problemi psichiatrici che si alimentava solamente di pasta con l’olio e che con questa scarsa alimentazione aveva sviluppato lo scorbuto. Anche soggetti che fanno una dieta vegetariana stretta possono avere una carenza di ferro, perché il ferro presente nei vegetali è spesso un ferro trivalente e non bivalente, quindi difficilmente assorbibile. Gli spinaci sono si ricchi di ferro ma questo ferro viene assorbito solo in minima parte, lo stesso discorso vale per i legumi. Perciò concludendo quando si presenterà da voi un paziente con carenza di ferro pensate prima alle cause più comuni e non alla atransferrinemia congenita, situazione che personalmente il prof non ha mai visto!!! Regolazione del ferro a livello intestinale - approfondimento L'intestino regola l'assorbimento del Ferro. In questo processo agiscono diverse proteine e la più importante è DMT1 che permette l'ingresso nella cellula del ferro bivalente. La riduzione del ferro è operata da un'altra proteina: la DcytB. Per essere mobilizzato e trasportato agli altri tessuti, il ferro, viene dapprima ossidato dall'efestina (negli enterociti) o dalla Ceruloplasmina (nelle altre cellule) e poi attraverso la ferroportina, viene portato fuori dalla cellula, dove si lega alla transferrina che ha la funzione di trasportarlo. La Transferrina viene captata dai vari tessuti attraverso dei recettori (TfR), si forma un endosoma, si libera ferro all'interno della cellula, e successivamente, il recettore torna in superficie. Nel 2000 è stato scoperto un peptide antimicrobico prodotto dal fegato, che ha un ruolo chiave nell'assorbimento del ferro: l'ormone Epcidina dall'inglese Hepcidin (da Hepatic Bacteriocidal Protein). Sembra strano che una proteina antimicrobica abbia un importante ruolo nell'assorbimento del ferro, ma i batteri per il loro sviluppo necessitano del ferro, che prendono dalle cellule attraverso particolari strutture, denominate siderofori. Quindi l'abbondanza di ferro nel nostro organismo favorisce le infezioni. L'epcidina agisce legando la ferroportina, che viene così degradata all'interno della cellula, e si blocca l'esportazione del ferro. Questo meccanismo agisce in particolare in 2 tipi di cellule: gli enterociti e i macrofagi, quindi l'epcidina agisce sull'assorbimento intestinale e sul rilascio dai macrofagi. Infatti bloccando l'esportazione, gli enterociti aumentano il loro contenuto in ferro, diminuendo così l'assorbimento, ed anche le perdite, dovute alla desquamazione delle cellule intestinali. Quando c'è un sovraccarico di ferro, oppure un infezione, il fegato aumenta la produzione di epcidina, che a sua volta diminuisce l'assorbimento a livello intestinale, e viene diminuito il rilascio dai macrofagi. Viceversa in stati carenziali di ferro, diminuisce la sintesi di epcidina, e viene aumentarto quindi l'assorbimento di ferro e il rilascio da parte dei macrofagi. Lezione 4 prof Di Raimondo Vi ho fatto vedere come ci sono queste tre possibili cause di anemia: - Perdita di sangue - Distruzione di globuli rossi - Ridotta produzione di globuli rossi Quindi come nella diagnostica delle anemia sia importante valutare: -volume globulare medio, che differenzia le anemie microcitiche, normocitiche e macrocitiche; -conta dei reticolociti , che permette di distinguere le anemie ipogenerative da quelle in cui c’è perdita o distruzione di globuli rossi; Abbiamo visto: - quali sono i sintomi dell’anemia, che ricordiamo sono sintomi legati non tanto all’entità dell’anemia quanto alla rapidità di insorgenza di questi valori; - che ci sono alcuni sintomi specifici per qualche anemia, tipo per esempio le ulcere alle gambe che sono tipiche dell’anemia falciforme; - l’importanza della morfologia nell’orientamento diagnostico delle anemie; E abbiamo iniziato a parlare delle anemie microcitiche. Si è visto come è distribuito il ferro nell’organismo, come si assorbe, quali sono le necessità di avere il ferro nell’organismo, quali sono le modalità di trasporto e di accumulo e quali sono le cause che aumentano le necessità di ferro e quali sono le cause di carenza di ferro. ANEMIE MICROCITICHE CARENZA DI FERRO - ANEMIA SIDEROPENICA Quindi vi ricordo che in caso di anemia da carenza di ferro la cosa più probabile è che il paziente abbia un eccesso di perdita di ferro: - nelle donne è verosimilmente legato alle perdite mestruali; - negli uomini è legata più spesso ad una lesione del tubo gastroenterico quindi ulcera peptica, emorroidi, neoplasia del colon, ernia iatale ; Meno frequenti sono le condizioni in cui il ferro manca per deficit di assorbimento, però vi dicevo che sempre di più l’utilizzo di inibitori di pompa favorisce questa carenza di ferro. Poi ci sono condizioni fisiologiche come la gravidanza e l’accrescimento; molto raramente un insufficiente apporto di ferro nei soggetti vegetariani stretti. Quindi quando manca il ferro cosa succede? Nella fase iniziale c’è una deplezione dei depositi di ferro (i quali si trovano soprattutto nel fegato ma anche nel midollo). Per cui se teoricamente facessimo un prelievo di midollo in una fase iniziale di carenza di ferro e andiamo a marcare il midollo con un colorante per il ferro, vedremmo che c’è una deplezione dei fisiologici depositi di ferro (è chiaro che non è pensabile andare a fare una biopsia del midollo per andare a cercare un inizio di carenza di ferro); -Riduzione della ferritina: quello che però possiamo andare a vedere è una riduzione di quello che è il marcatore del ferro di deposito, cioè la ferritina (la ferritina che troviamo nel sangue è proporzionale alla ferritina presente nei tessuti, per cui è uno specchio della carenza di ferro dei depositi.) Quindi il dosaggio della ferritina ci dice che ci sono i depositi di ferro depauperati, e questo è uno dei primi segni della carenza di ferro, perché appunto quando l’organismo ha bisogno di ferro se lo va a pigliare dove c’è il deposito. -Aumento della transferrina: un altro segno della carenza di ferro è l’aumento della transferrina, perché vi dicevo la transferrina è il trasportatore del ferro, che viene definito anche TIBC (total iron binding capacity) che indentifica l’avidità dell’organismo per il ferro, quindi è chiaro che in carenza di ferro c’è un aumento della transferrina. Gli altri parametri nella fase iniziale di questa condizione di sideropenia sono normali: - Sideremia normale: il ferro circolante rappresentato dalla sideremia è normale; - Percentuale di saturazione della transferrina è normale; - Emocromo è normale. In una fase un po’ più spinta inizia l’eritropoiesi con deficit di ferro, in questo caso: all’abbassamento dei valori di ferritina e all’aumento dei valori di transferrina, fanno seguito pure -abbassamento dei valori di sideremia, cioè del ferro circolante, -ridotta saturazione della transferrina; ancora una volta qua l’emocromo è normale. Nella fase più spinta inizia l’anemia, quindi i depositi di ferro sono del tutto depleti: -transferrina aumentata; -ferritina bassa; -sideremia bassa; -saturazione della transferrina bassa; -sideroblasti nel midollo sono bassi; - e qui inizia l’abbassamento dei valori di emoglobina che passa ad es da 15 a 13 ed inizia l’ANEMIA MICROCITICA, per cui i globuli rossi saranno microcitici (piccoli) e ipocromici (poco colorati). Infine nella condizione più evidente non solo c’è tutto quello che abbiamo visto prima, ma in più i -valori di emoglobina si abbassano di molto (qui vedete 6 gr di emoglobina che è un valore assolutamente basso, meno della metà dei valori normali); -e c’è anche una forte riduzione del volume globulare medio. Quindi questa è una progressione della condizione di carenza di ferro: da una deplezione di depositi si arriva ad una condizione di anemia microcitica conclamata. Emocromo classico di un paziente con anemia da carenza di ferro : dove vedete il voume globulare medio è basso (L indica low: molti contaglobuli automatici danno un asterisco ove i valori sono alterati) Globuli rossi bassi:4.280.000; una bassa emoglobina 9.7; un basso ematocrito 29 e un basso MCV (questo vuol dire che i globuli rossi sono piccoli); questo si tira dietro anche l’MCH e MCHC e abbiamo invece un alto RDV: poi vedremo cosa significa. Vi ricordo che l’anemia microcitica è caratterizzata anche da - cheilite angolare (fissurazioni agli angoli della bocca) - glossite (lingua disepitelializzata), - coilonichia(unghie a cucchiaio), che sono abbastanza tipiche della carenza di ferro Oltre alla carenza di ferro ci può essere anche l’eccesso di ferro ECCESSO DI FERRO L’eccesso di ferro è dovuto soprattutto a delle condizioni ereditarie, quali per esempio l’emocromatosi o la β-talassemia. In questo caso c’è un eccesso di assorbimento di ferro, ci può essere un aumento dell’intake (ad es per una terapia parenterale con ferro per un lungo periodo di ferro o cmq eccessiva) oppure eccesso di trasfusioni Aumento modesto del carico di ferro: tutte le malattie epatiche, una rara malattia che si chiama Porfiria cutanea tarda e altre condizioni più rare quali l’atransferrinemia e l’aceruloplasminemia. Emocromatosi ereditaria L’emocromatosi ereditaria è una condizione congenita nella quale c’è un graduale e progressivo accumulo di ferro, talmente lento che la manifestazione della malattia si verifica alla 5° decade di vita o anche più tardi. Quindi sono pazienti con condizioni congenite che si manifestano a 50- 60 anni. Chiaramente è una malattia che colpisce soprattutto gli uomini, perché le donne avendo le mestruazioni perdono ferro con le mestruazioni ed evitano l’accumulo di questo ferro. I sintomi di questa malattia sono abbastanza vaghi: sensazione di stanchezza, perdita di libido, perdita di peso,artralgia. Solo più tardivamente ci sono manifestazioni da accumulo di ferro che sono le atropatie perché il ferro si accumula nelle articolazioni; la iperpigmentazione (colorito bronzino) della pelle; soprattutto la compromissione cardiaca perché il ferro si accumula molto nel cuore. La diagnosi come si fa? Si fa quando si vede per esempio un paziente diabetico, il cosiddetto diabete bronzino, che non è altro che un accumulo di ferro nel pancreas, che causa una distruzione nel pancreas (quindi diabete) associato ad un colorito bronzino della pelle. Il paziente con artrite, insufficienza gonadica che può essere presente già alla nascita, ma soprattutto c’è una compromissione epatica (il ferro si accumula soprattutto nel fegato) e tutto questo si accompagna a colorito particolarmente scuro della cute con dei segni laboratoristici di accumulo di ferro; Due parametri da considerare nella diagnosi di emocromatosi: -in particolare deve essere presente una concentrazione di ferritina superiore a 1000; -e una saturazione della transferrina superiore all’80%; Trattamento: questa malattia si tratta facendo i salassi; adesso ci sono i nuovi chelanti del ferro (il vecchio chelante era la deferoxamina ) che sono promettenti terapie future, ma oggi si fanno ripetuti salassi. La diagnosi viene sospettata con la valutazione di ferritinemia e saturazione della transferrina, ma poi per valutare il difetto genetico si fanno indagini genetiche per trovare il tipo di mutazioni responsabili delle alterazioni: mutazioni di quelle proteine come la ferro cortina (vedi lezioni precedenti) che servono a regolare l’ingresso del ferro a livello dei villi intestinali nel sangue. Bastano delle alterazioni puntiformi di queste proteine per determinare un eccesso di passaggio nel sangue da parte del tubo gastroenterico e giustificano appunto questo tipo di patologia. Ci sono una gamma di condizioni che vanno dall’omozigosi all’eterozigosi e a seconda della proteina che è interessata in questo tipo di trasporto hanno diverse manifestazioni. Da ricordare ancora una volta che l’emoglobina è formata da un tetrametro di 4 catene globiniche più 4 gruppi eme, e in questo è presente Fe più la protoporfirina IX (queste sono nozioni di base che dovete tenere a mente per capire anche la patologia). Vi ricordo anche che l’emopoiesi è differente nel periodo embrionale, fetale e post natale: nel periodo embrionale l’emopoiesi è affidata al sacco vitellino, nel periodo fetale al fegato ed infine è affidata al midollo osseo. Ma la costituzione delle catene globiniche è differente nei vari periodi, per cui nel periodo embrionale e fetale ci sono delle catene globiniche che poi non sono più presenti dopo la nascita e nel periodo fetale cominciano ad essere prodotte le emoglobine che poi saranno largamente rappresentate nell’adulto che sono la globina α e la globina β; nel periodo fetale è preponderante rispetto alla β la globina γ che è presente durante tutto il periodo fetale per poi ridursi alla nascita e quasi scomparire nell’adulto. Quindi questo cross over tra la globina β e la globina γ dal periodo fetale al periodo dell’adulto è molto importante anche per capire alcuni aspetti delle patologie. Quindi nel periodo prenatale avremo questo tipo di emoglobina gower 1, gower 2, portland, emoglobina H. Nel periodo post natale fondamentalmente c’è prevalentemte Hb A (α2β2), molto meno Hb A1c (α2β2 glicata) e Hb A2 (α2δ2) e ancor meno Hb F (α2γ2). Quindi tutto questo sistema può essere alterato in patologia, in delle condizioni definite emoglobinopatie e la più classica è la talassemia. Quindi ci possono essere alterazioni della struttura dell’emoglobina: - anomala polimerizzazione (HbS nell’anemia falciforme) - anomala cristallizzazione (HbC) - emoglobine instabili - emoglobine con aumentata affinità per l’ossigeno - emoglobine con diminuita affinità per l’ossigeno I difetti invece quantitativi nella produzione delle catene emoglobiniche sono le talassemie, che si differenziano a seconda del deficit della globina: se il deficit interessa la produzione di catene α saranno α-talassemie; se il deficit interessa le catene β si chiameranno β-talassemie Poi ci può essere la persistenza dell’emoglobina fetale (che in condizioni normali come visto è assolutamente assente). Poi ci possono essere delle altre condizioni in cui c’è un’alterazione della forma, della struttura dell’emoglobina, per condizioni acquisite, e la più famosa è la metaemoglobina. La più importante di queste patologie è sicuramente la Talassemia Talassemia La Talassemia è la conseguenza di un difetto di produzione delle catene globiniche, quindi: - se manca o è alterato il gene che produce le α catene ci sarà l’α-Talassemia; - se è alterato il gene che produce le β catene ci sarà la β-Talassemia. Patogenesi Abbiamo visto che le catene α e β sono in equilibrio, se ne manca una, ci sarà uno squilibrio, cioè ci sarà una discrepanza tra la quantità di catene α e la quantità di catene β, cioè l’una o l’altra saranno presenti in maggior quantità. Nel caso della β-talassemia, quindi ci sarà un eccesso di catene α, e all’interno degli eritroblasti si formeranno degli aggregati di catene α; questi aggregati tendono a precipitare rendendo così gli eritroblasti anomali. Gli eritroblasti anomali vengono rapidamente distrutti e quindi si parla di eritropoiesi inefficace: cioè l’eritropoiesi c’è ma non arriva a maturazione per distruzione rapida e precoce di questi eritroblasti. Tutto questo comporta chiaramente un’anemia, e l’anemia comporta un aumento dell’assorbimento intestinale di ferro e quindi una condizione di emocromatosi secondaria (abbiamo visto come l’emocromatosi è da accumulo di ferro). Questi pazienti che sono appunto anemici e hanno un elevato turnover cellulare in cui l’eritrone è molto espanso, cioè c’è un’elevata produzione di globuli rossi, ma questi sono anomali dunque rapidamente distrutti e quindi l’organismo essendo anemico aumenta l’assorbimento di ferro; l’assorbimento di ferro viene peggiorato dalla terapia trasfusionale e quindi c’è un sovraccarico di ferro. Una delle problematiche più grosse in questi pazienti è proprio il sovraccarico di ferro che provocherà un’emocromatosi per deposito del ferro a carico del cuore, del fegato, delle isole pancreatiche, delle ghiandole endocrine: in pratica dappertutto, ubiquitario. Non soltanto gli eritroblasti ma anche gli eritrociti sono anomali, e anche qua gli aggregati insolubili di catene α precipitano e vengono rapidamente riconosciuti dalla milza come anomali, quindi la milza li distrugge, quindi c’è quest’emolisi che contribuisce al’anemia microcitica. L’anemia microcitica provoca ipossia, quindi l’organismo risponde aumentando la produzione di eritropoietina, quindi aumentando l’eritropoiesi: vi dicevo che c’è un’eritropoiesi particolarmente attiva in questi pazienti e questo determina non solo un aumento del tessuto midollare nel midollo osseo (che determinerà quelle deformazioni scheletriche tipiche della Talassemia, proprio da espansione del midollo osseo), ma ci sarà anche un’eritropoiesi extramidollare, in particolare fegato e milza riprendono la loro funzione che avevano nella vita fetale e cominciano anche loro a produrre globuli rossi: è un’ eritropoiesi assolutamente inefficace, insufficiente, e questo determina un ulteriore ingrandimento del fegato e della milza; la milza è ingrandita perché fa il suo mestiere nell’emolisi dei globuli rossi alterati. Tutto questo configura il quadro clinico del paziente talassemico. Ci sono diversi quadri clinici che dipendono dall’entità del difetto genetico. Un soggetto normale avrà: 2 geni che codificano per le catene globiniche β; 4 geni che codificano per le catene globiniche α. Egli avrà soprattutto Hb A(α2β2), bassa percentuale (fino al 3%) di emoglobina A2 (α2δ2) e una piccola quota di emoglobina fetale (minore dell1%). β-Talassemie Partiamo dalle β-TALASSEMIE che sono più semplici e tra queste si distinguono tre condizioni a seconda del difetto genetico: - talassemia minor, - intermedia - talassemia maior (morbo di Cooley) → se mancano ambedue i geni che codificano per le catene β, avremo l’assoluta mancanza di cateneβ e quindi la TALASSEMIA MAJOR o MORBO DI COOLEY → se manca un solo gene che codifica per le catene β, avremo la TALASSEMIA INTERMEDIA → se invece c’è il parziale silenziamento di un geneβ e l’altro funziona, avremo la TALASSEMIA MINOR. La TALASSEMIA MINOR è caratterizzata soltanto da un lieve aumento dell’Hb A2; si fa un esame che si chiama elettroforesi dell’emoglobina, per cui queste varie Hb migrano in un campo elettrico a seconda del peso, e si può evidenziare dove migrano queste Hb e fare una valutazione quantitativa delle diverse emoglobine. Per cui se un paziente ha un aumento dell’Hb A2 che in condizioni normali è minore del 3%, quella è un’indicazione che il paziente è portatore di talassemia, una condizione molto frequente dalle nostre parti ed è la modalità per fare prevenzione della talassemia: ormai è un esame che viene fatto nelle scuole, a tutti gli sposini che vogliono un figlio per evidenziare lo stato di portatore di talassemia. Questi soggetti sono soggetti normali che hanno soltanto molto spesso una riduzione dell’MCV (perché la talassemia è un’anemia tipicamente microcitica), ma attenzione perché non è sempre presente questa riduzione dell’MCV, ci sono portatori per i quali soltanto facendo l’elettroforesi dell’emoglobina si vede che sono affetti da talassemia minor, per il resto non c’è nessun altra anomalia e nella stragrande maggioranza dei casi hanno valori di emoglobina vicini alla norma. Il quadro della TALASSEMIA INTERMEDIA è invece un quadro clinico evidente ove il paziente oltre ad avere un aumento dell’HbA2 , ha anche un aumento dell’ HbF, che è una sorta di aumento compensatorio rispetto alle altre Hb e in questo caso il paziente è anemico e a volte ha bisogno di un apporto trasfusionale, senza però arrivare ai livelli del morbo di Cooley. α-Talassemie Più complessa invece è l’α-TALASSEMIA, il concetto comunque è sempre il medesimo: - se mancano tutti i geni α, ci sarà una condizione severa - se invece ha soltanto un difetto parziale di questi geni, la condizione non sarà clinicamente evidente Quando mancano tutti i geni α, si avrà la cosiddetta idrope fetale, una condizione è incompatibile con la vita. La condizione di α-talassemia non è che è più grave della β-Talassemia, è semplicemente che nella β-talassemia si può formare l’Hb F, perché sono presenti i geni α, mentre nell’α-Talassemia non si può formare neanche l’Hb F, quindi il feto manca di quell’HbF (α2γ2), perché non ci sono catene α. Quindi il bambino con la β talassemia, fino a che è nella sua condizione fetale non ha grossi disturbi perché le catene β si producono soltanto nel periodo immediatamente prima della nascita e servono poi dopo la nascita, ma dopo 3-6 mesi sono indispensabili le cateneβ; per cui il bambino con βtalassemia manifesta la sua condizione di anemia dopo 6 mesi dalla nascita, mentre il bambino con α Talassemia muore prima perché non può formare neanche l’HbF. - quando mancano 3 geni α si ha una certa Hb Barts? che è formata soltanto da catene β ed è una condizione patologica importante; - nelle altre condizioni, in cui mancano 2 geni o un gene α, invece non sono condizioni cliniche e sono del tutto paragonabili alla β Talassemia minor, assolutamente silenti sul piano clinico. Sintomi e Segni Quindi le manifestazioni della β Talassemia sono manifestazioni scheletriche che sono legate soprattutto all’espansione del midollo. Questi pazienti hanno una cardiomiopatia importante che è dovuta in parte all’anemia severa (ricordate quando abbiamo detto dei sintomi delle anemie, ci possono essere compromissioni cardiache legate all’eccessiva gittata, all’eccessiva diluizione di questo sangue, ma in parte è legata all’eccesso di ferro che va a depositarsi nel cuore). Inoltre c’è un difetto di accrescimento, in parte legato all’anemia, in parte legato all’accumulo di ferro nelle ghiandole endocrine; c’è epatomegalia, in parte legata all’ematopoiesi extramidollare, in parte legata all’accumulo di ferro nel fegato. § Questi sono dei quadri clinici di talassemia, vedete il polimorfismo di questi elementi: emazia a bersaglio, ellissocita, emazia a lacrima…ci sono diverse forme di questi globuli rossi. Nella β-talassemia major come vedete nel sangue periferico la presenza di eritroblasti, questa cellula qua ha un citoplasma identico a quello di un globulo rosso, ma c’è ancora il nucleo; questa è una sezione del cranio di un paziente talassemico, ovviamente un reperto autoptico: vedete come c’è una grossa espansione generale di tutta l’emopoiesi e quindi del midollo osseo; queste sono colorazioni del ferro: vedete tutte queste punteggiature blu scure sono tutti depositi di ferro nel muscolo cardiaco, e questa è una cardiomiopatia con cardiomegalia a causa dell’anemia e dell’eccesso di ferro, che adesso si può evidenziare anche con metodiche di Risonanza Magnetica: questa è una risonanza magnetica del cuore e come vedete questo cerchio nero è tutto accumulo di ferro in questi paziente. Trattamento: i pazienti talassemici vanno trattati soltanto se sintomatici. Quindi se un paziente è solo portatore di talassemia (α-minor o β-minor) non c’è da fare alcun trattamento terapeutico ma solo la prevenzione genetica, cioè far conoscere a quel soggetto che se fa un figlio con un altro portatore di talassemia c’è il rischio di fare un figlio malato. Invece i pazienti talassemici vanno trattati con: - trasfusioni di sangue per mantenere alti i valori di Hb; - si fa terapia chelante, qui rappresentata dalla desferrossamina(?) ma vi dicevo esistono nuovi chelanti del ferro che sono particolarmente efficaci e molto più comodi da somministrare rispetto alla desferrossamina(?). - va fatta la splenectomia, perché appunto in questi pazienti la milza molto grande serve da distruttore di globuli rossi (ricordatevi che quando si fa una qualunque splenectomia, questa deve essere accompagnata da un vaccino anti-pneumococcico, perché i pazienti splenectomizzati corrono il rischio di avere infezioni letali da germi capsulati, soprattutto pneumococco.). Poi è utile una supplementazione di acido folico, vitamina C, vitamina E per supportare questa eritropoiesi aumentata. Quindi fino adesso tra le anemie microcitiche sono state viste l’anemia da carenza di ferro e la talassemia, un’altra condizione che può essere considerata un’anemia microcitica (ma non sempre è microcitica, più spesso è normocitica), è la cosiddetta anemia delle malattie croniche. ANEMIA DELLE MALATTIE CRONICHE Questa è una condizione che non viene spesso considerata, è un po’ negletta (trascurata) come diagnostica ma in realtà è un’anemia molto frequente. E’ un’anemia tipicamente ipoproliferativa, quindi avremo una conta di reticolociti bassa o relativamente bassa e si accompagna a infezioni, neoplasie, malattie autoimmuni e a volte anche traumatismi continui; è l’anemia più comune nelle corsie di ospedale! Però è un’anemia molto modesta, generalmente non si arriva mai a valori molto bassi di Hb; usualmente i globuli rossi sono normocitici o lievemente microcitici e c’è una variabile ipocromia. La caratteristica peculiare di questa anemia è il fatto di avere una sideremia bassa nonostante i depositi di ferro siano normali o addirittura aumentati. Quindi avremo sideremia bassa e ferritina normale o aumentata. Diverse sono le condizioni che si possono associare: condizioni infettive, infiammatorie, molte neoplasie e questo deriva dal fatto che tutte queste condizioni sono caratterizzate dalla iperproduzione di alcune citochine: IL-1 , TNF, IL-4 che alterano il normale metabolismo del ferro, riducono la produzione di globuli rossi e riducono anche la risposta all’eritropoietina. Da ricordare che una delle molecole più importanti per regolare il traffico del ferro dal lume intestinale alla cellula, all’eritroblasto, è una molecola che si chiama epcidina, che serve anche a regolare questo traffico del ferro nel macrofago, quindi questa molecola è estremamente importante nella regolazione del traffico del ferro. In tutte queste anemie c’è un aumento dell’epcidina che causa una condizione di blocco sia dell’assorbimento del ferro, sia della redistribuzione del ferro dal sistema reticolo-endoteliale al sangue → quindi questa condizione determinerà una bassa sideremia (cioè bassa quantità di ferro circolante), ma un accumulo di ferro nei depositi e un ridotto assorbimento di ferro. Quindi la diagnostica va fatta escludendo che si tratti di un’altra forma di anemia, e confermando che si tratta di una anemia ipoproliferativa, quindi bassi reticolociti con questa diversa distribuzione del ferro, sideremia bassa ma ferritina alta e tutto questo inquadrato in un ambito di una malattia cronica. Riassunto: i reticolociti non saranno aumentati, la bilirubina sarà normale, LDH sarà normale, l’aptoglobina non sarà ridotta; vi sarà una bassa sideremia, una bassa trasferrina, una bassa saturazione della trasferrina, ma la ferritina sarà normale o aumentata; e ovviamente troveremo tutti gli indicatori di infiammazione quali fibrinogeno aumentato, ceruloplasmina aumentata, proteina c aumentata ecc…. spesso ci sarà anche una bassa concentrazione di albumina e un aumento della ves. In genere questi pazienti non hanno bisogno di terapia specifica per l’anemia, ma basta togliere la causa infiammatoria di base per far guarire questa anemia. In alcune condizioni è necessario fare la terapia con l’eritropoietina ricombinante che aumenta la produzione di globuli rossi e in alcune patologie croniche difficili da guarire, questa somministrazione può essere utile nel migliorare il quadro clinico. Quindi è importante fare uno schema molto esemplificativo e molto importante che valuta i tre parametri che dobbiamo utilizzare nella diagnostica delle anemie microcitiche, cioè la sideremia, la transferrina e la ferritina e le valuta nelle condizioni che abbiamo visto prima cioè le anemie sideropeniche, le talassemie e le malattie secondarie a malattia cronica. → Nella sideropenia, anemia sideropenica, abbiamo visto che c’è una deplezione del ferro, e quindi ci sarà una sideremia bassa (ferro circolante basso), ferritina bassa (i depositi di ferro sono bassi), ma la trasferrina alta (l’avidità del ferro per l’organismo aumenta). → Nelle anemie secondarie a malattie croniche invece c’è sideremia bassa, perché il ferro non viene mobilizzato, rimane lì dov’è; quindi avremo in questo caso la ferritina alta, e anche molto spesso la transferrina bassa. Quindi vedete come queste due condizioni sono caratterizzate dall’avere entrambe la sideremia bassa, ma c’è una differenza con la ferritina e la transferrina. Questo perché è importante? Perché molto spesso si vedono pazienti anemici e per vedere se sono anemici si fa soltanto la valutazione della sideremia, ma soltanto con la valutazione della sideremia non riusciamo a fare diagnosi differenziale tra queste due condizioni, perché ambedue le condizioni hanno la sideremia bassa! La diagnosi differenziale la posso fare soltanto facendo anche la valutazione della ferritina e della transferrina, che saranno assolutamente differenti. Per l’anemia microcitica, vi dicevo prima, ovviamente si deve fare diagnosi differenziale tra la condizione di sideropenia e la condizione di portatore di talassemia. Ma qui è abbastanza facile, perché è vero che sono ambedue anemie microcitiche, però nel caso della talassemia tutti i marcatori del ferro saranno aumentati, quindi avremo aumento della sideremia, aumento della ferritina e avremo una transferrina normale o aumentata; questi dati possono essere normali oppure aumentati, ma non avremo la carenza che abbiamo nell’anemia sideropenica. Per cui più difficile è la diagnosi differenziale tra condizione di sideropenia e condizione di malattie croniche piuttosto che la diagnosi differenziale tra condizione di sideropenie e talassemie. Però ricordatevi che tutte e tre le condizioni possono essere anemie microcitiche: quindi l’anemia microcitica è possibile che sia anemia sideropenica, è possibile che sia talassemica e molto meno frequentemente ma è anche possibile che sia un’anemia secondaria a malattie croniche. In quest’ultimo caso il ferro non viene mobilizzato quindi sarà presente nei depositi, non sarà presente il ferro circolante, nel caso dell’anemia sideropenica sono invece depletati i depositi di ferro, nel caso delle talassemie c’è in genere un accumulo di ferro e non una deplezione. Si diceva prima dell’importanza dell’ RDV, se ricordate l’emocromo che possiamo avere in un paziente con anemia sideropenica, accanto ai parametri sottolineati in precedenza c’è l’RDV che nel caso specifico è aumentato. Cos’è l’RDV? È l’indice di anisocitosi: l’acronimo sta per Red Cell Distribuition Volume, cioè l’ampiezza di distribuzione dei globuli rossi. I globuli rossi hanno un volume medio ma chiaramente non sono tutti uguali, questo volume si distribuisce in una gaussiana e questa gaussiana è rappresentata dall’RDV. Nell’ambito delle anemie microcitiche, ci sono le anemie microcitiche congenite, come la talassemia, dove i globuli rossi sono piccoli tutti quanti allo stesso modo proprio perché sono congenite. Viceversa, l’anemia microcitica da carenza di ferro è una condizione acquisita quindi il globulo rosso è programmato per essere di un certo volume ma siccome gli manca il ferro si riduce il suo volume; ma la capacità di resistere a questa condizione è diversa tra un globulo rosso e l’altro, quindi possiamo dire che ogni globulo rosso ruba il ferro al suo vicino, e chi è più forte riesce a non essere microcitico per cui questa distribuzione della gaussiana è più ampia nelle anemie microcitiche perché c’è una maggiore variabilità nell’essere microcitici di questi globuli rossi. E quindi una delle caratteristiche differenziali tra anemia microcitica da carenza di ferro e anemia microcitica da talassemia è proprio nell’RDV: dove tipicamente nella carenza di ferro c’è un RDV ampio, più ampio di quello che troviamo nel paziente con talassemia. E quindi l’RDV può essere utilizzato come primo elemento di diagnostica differenziale in un paziente con anemia microcitica. Se avete un paziente con anemia microcitica e non avete altri esami a disposizione, cioè non avete ferritina, sideremia, né elettroforesi dell’Hb (che vi dice se quel paziente è talassemico oppure no), una prima valutazione può essere fatta con l’RDV: se l’RDV è più ampio del normale è molto probabile una sideropenia, e se invece l’RDV è normale è più probabile che sia un paziente con uno stato di portatore sano di talassemia. Quindi ecco il facile algoritmo della diagnostica delle anemie (ovviamente ci deve essere un’anemia Hb<14gr nell’uomo; Hb<12 gr nella donna), in questo caso occorre esaminare l’MCV: - se l’MCV è ridotto di tratterà di un’anemia microcitica (e abbiamo visto quali sono le cause di anemia microcitica: talassemia, anemia sideropenica e più raramente l’anemia da flogosi croniche) -se l’MCV è normale chiaramente parliamo di anemia normocitica o macrocitica --se è macrocitica cioè se ha un volume globulare medio più elevato vedremo successivamente… --se è normocitica la cosa da fare è la conta dei reticolociti: → se i reticolociti sono elevati, l’anemia può essere soltanto emolitica o emorragica → se i reticolociti sono bassi, l’anemia può essere ipoproliferativa o da eritropoiesi inefficace Questi due parametri sono quindi fondamentali nella diagnostica delle anemie, l’MCV e la conta dei reticolociti perché ci permettono di differenziare le varie condizioni dell’anemia. ANEMIE MACROCITICHE Anche le anemie macrocitiche sono abbastanza limitate, fondamentalmente distinguiamo anemie megaloblastiche e non megaloblastiche: Megaloblastiche: mcv >120 Non megaloblastiche: 100 < mcv <120 * anche se queste sono delle distinzioni non del tutto precise. MEGALOBLASTICHE: Nelle anemie in cui c’è un aumento del volume globulare medio superiore a 100 sono le anemie da carenza di B12 e di acido folico. Ci sono però alcune sindromi pre-leucemiche ed epatopatie croniche che possono determinare questo aumento oppure infine ci sono pazienti trattati con antimetaboliti (in pratica è come se mancasse la B12 o l’acido folico) tipo il metotrexate in cui appunto c’è aumento dell’mcv. L’anemia megaloblastica è quindi un’anemia da carenza di B12 e di acido folico e in alcune parti del mondo è la seconda più comune causa di anemia. L’anemia da carenza di B12 è una condizione multisistemica perché non è che la B12 e l’acido folico sono utili soltanto per fare i globuli rossi, ma sono utili anche per altri tessuti, in particolare il tessuto nervoso necessita di vit B12. L’anemia megaloblastica è caratterizzata dall’essere un’anemia macrocitica, ed è non soltanto anemia, ma più spesso c’è una pancitopenia. Molti casi di anemia megaloblastica sono configurati in quella che viene definita anemia perniciosa: condizione in cui a causa di un processo autoimmune, cioè di anticorpi contro le cellule parietali gastriche si viene a creare un’atrofia della mucosa dello stomaco, che determina una carenza nell’assorbimento di vitB12; sapete infatti che la vit. B12 per essere assorbita necessita del fattore intrinseco che viene prodotto dalle cellule parietali gastriche. In assenza di questo fattore intrinseco la vit.B12 non può essere assorbita, in particolare l’assorbimento avviene nell’ileo, non avviene nello stomaco, ma se lo stomaco non produce il fattore intrinseco non viene assorbita. Cause di anemia megaloblastica: - malnutrizione (chiaramente se non si ingerisce vitB12); - mancanza di fattore intrinseco, cioè l’anemia perniciosa; - il paziente gatrectomizzato non ha fattore intrinseco, oppure il paziente a cui manca il distretto dove viene assorbita (ileo); - malattie infiammatorie dell’intestino o malassorbimenti; Patogenesi: mancando la vit. B12 o i folati si ha un difetto della sintesi di DNA, quindi la maturazione degli eritroblasti viene ritardata, tutto questo fa aumentare il volume del globulo rosso per cui si ha una macrocitosi che però contiene una quota normale di emoglobina, quindi la colorazione del globulo rosso sarà normocromica e non ipocromica come ad esempio nel caso della carenza di ferro; quindi c’è basso numero di globuli rossi, basso numero di globuli bianchi e spesso c’è anche un basso numero di piastrine. Questo è l’emocromo tipico di un paziente con l’anemia perniciosa, dove appunto c’è una riduzione dei valori di emoglobina, ma con un volume globulare medio molto alto (117). L’anemia megaloblastica si caratterizza inoltre per anomalie a carico dei globuli bianchi, in particolare c’è un segno che è la plurinuclearità, plurilobularità del neutrofilo che è un segno abbastanza specifico della carenza di B12; Quindi per definire un po’ la diagnosi differenziale delle anemie macrocitiche: - nell’Anemia Megaloblastica il volume globulare >110, i globuli bianchi spesso sono diminuiti, le piastrine diminuite, c’è l’ipersegmentazione dei neutrofili e ovviamente andando a fare un dosaggio di B12 e di folati ovviamente troveremo una riduzione (più spesso della B12, è più rara la carenza di folati). - Altre condizioni cui si associano anemie macrocitiche sono le Anemie Associate a Epatopatie Croniche. In questo caso usualmente il volume globulare è un pochetto più basso e non c’è riduzione dei globuli bianchi (o perlomeno ci può essere ma per altri motivi perché spesso lì c’è un ipersplenismo, e quindi lì c’è diminuzione dei globuli bianchi e delle piastrine), ma chiaramente se andiamo a dosare folati e B12 li troviamo normali. - Un’altra condizione di macrocitosi sono alcune Sindromi Preleucemiche dove anche qui sono spesso diminuiti globuli bianchi e piastrine e anche qui non c’è ipersegmentazione dei nuclei dei neutrofili e anche la B12 e acido folico sono normali. Quindi queste sono le condizioni più frequenti di anemia macrocitica: anemia megaloblastica, anemia delle epatopatie croniche e la sindrome preleucemica. ANEMIE NORMOCITICHE Quando abbiamo un’anemia normocitica dobbiamo considerare innanzi tutto il valore dei globuli bianchi e delle piastrine, perché se valori di globuli bianchi e piastrine sono alterati, dobbiamo considerare la possibilità di fare una valutazione del midollo, perché ci possono essere delle Patologie Midollari (visto che c’è compromissione di globuli bianchi e piastrine), quindi dobbiamo pensare che sia una compromissione di tutto il midollo, non soltanto dell’eritropoiesi. Se invece i globuli bianchi e le piastrine sono normali, dobbiamo pensare prima di tutto all’Anemia da Malattie Croniche; in questo caso è importante il conteggio dei reticolociti perché appunto ci permette di differenziare - se sono aumentati le emorragie emolitiche, - se non sono aumentati anche qua bisogna considerare se c’è un’anamnesi positiva per malattie croniche abbiamo fatto la diagnosi, andiamo a vedere com’è la ferritina, sideremia ecc…, se invece non c’è questa malattia cronica bisogna andare a vedere se ci sono delle anomalie midollari che giustificano questa condizione di anemia. § Introduzione alle ANEMIE EMOLITICHE CONGENITE Ricordiamo che i globuli rossi hanno una vita di 120 gg e che richiedono per funzionare un’integrità della struttura della membrana e del citoscheletro e un’energia sufficiente a far funzionare le pompe sodio-potassio; se c’è un’alterazione in una di queste condizioni si viene a creare l’emolisi. Quindi il globulo rosso è molto semplice come cellula, ha bisogno soltanto della sua integrità di membrana e di un minimo di energia. Questo vale per introdurre il discorso delle Anemie emolitiche congenite. Il metabolismo del globulo rosso è molto semplice: è legato alla glicolisi e ha necessità di ATP; questo metabolismo è basato su un enzima che è la glucosio-6-P deidrogenasi, un enzima cruciale nel metabolismo della cellula: in genere i radicali dell’ossigeno producono perossido di ossigeno che è detossificato dal glutatione. Questo sistema è importante perché tutto basato sul ruolo fisiologico della glucosio-6-P deidrogenasi, che serve a mantenere in vita il globulo rosso e proteggerlo dall’azione tossica dei superossidi per cui la mancanza di questo enzima è la causa maggiore di emolisi a causa dello stress ossidativo; e siccome il globulo rosso non ha nucleo non può produrre altro enzima, quindi se c’è n’è poco, non ne può produrre ancora quindi va incontro allo stress ossidativo. La membrana del globulo rosso è abbastanza complessa perché formata da un bilayer di fosfolipidi di membrana che sono molto ben connessi tra di loro; alcune proteine importanti sono la banda 3, l’anchirina e altre proteine meno importanti. Tutte le condizioni che provocano un’alterazione di queste proteine o del bilayer fosfolipidico daranno un’emolisi. Emolisi L’emolisi può essere intravascolare o extravascolare: - Emolisi Intravascolare significa che è un’emolisi che avviene per un problema legato al globulo rosso; - Emolisi extravascolare quando c’è qualcosa che agisce dall’esterno del globulo rosso. In generale l’EMOLISI INTRAVASCOLARE determina la liberazione di emoglobina libera, la quale nel sangue si complessa all’aptoglobina. Poi questo complesso viene trasportato al sistema reticolo-endoteliale e lì viene fagocitato. Per cui un segno di emolisi è di trovare l’aptoglobina bassa. Il paziente con l’emolisi avrà un LDH alto, i reticolociti alti. Quindi se dobbiamo andare a vedere se un paziente ha avuto un’emolisi, quello che dobbiamo andare a studiare sono: reticolociti, LDH, aptoglobina, bilirubina indiretta ridotta. Ma esistono delle condizioni in cui tutto questo è transitorio, il paziente ha soltanto un episodio di emolisi, seguito a distanza da un altro episodio di emolisi: tutto questo avviene nell’Emoglobinuria Parossistica Notturna in cui gli episodi di emolisi sono appunto parossistici, il grosso delle emolisi è occasionale. In questo caso se noi vediamo il paziente una settimana dopo che ha avuto un’emolisi, è chiaro che avrà ricostituito i valori di aptoglobina, quindi non troveremo più l’aptoglobina ridotta; non avrà più l’LDH. Però con l’emolisi succede che l’emoglobina libera nel sangue passa il filtro renale, supera la soglia renale di escrezione e va a depositarsi a livello dell’epitelio del tubulo renale, il quale è sempre sottoposto a una dismissione delle urine per cui nelle urine troviamo costantemente cellule dell’epitelio del tubulo renale: andando a colorare queste cellule con il colorante per il ferro, se c’è stata una emolisi anche diversi giorni prima, allora troveremo un aumento di ferro in queste cellule del tubulo renale che sono dismesse nelle urine e questa è la famosa emosiderinuria. Quindi l’emosiderinuria è una valutazione del ferro nelle cellule del sedimento urinario e a volte è un parametro utile per andare a valutare una pregressa emolisi. Nell’ EMOLISI EXTRAVASCOLARE, il meccanismo è lo stesso ma la quantità di emoglobina libera presente nel plasma è minore rispetto ad un’emolisi intravascolare e quindi la riduzione dell’aptoglobina sarà presente ma non sarà così spiccata come nel caso dell’emolisi intravascolare. Ma la diagnostica dell’anemia è fatta da: aumento dell’LDH (che è molto sensibile ma non specifico perché qualunque distruzione cellulare provoca un aumento dell’LDH); aumento della bilirubina indiretta ( anche questo qua non specifico, perché ci sono altre condizioni che la aumentano, ad es. sindrome di Gilbert è una condizione nella quale c’è un difetto nella glicurunazione della bilirubina; ma quella non è una malattia, è una condizione parafisiologica) ; aumento della conta dei reticolociti (è il parametro più importante che ci dice che c’è un’anemia emolitica); la riduzione dell’aptoglobina (perché si lega all’emoglobina libera), e l’emosiderina urinaria (però l’esame viene fatto in condizioni particolari). Per differenziare l’emolisi intravascolare ed extravascolare: Nell’emolisi intravascolare spesso troviamo la presenza di schistociti, cioè frammenti di globuli rossi che indicano che c’è stata una distruzione di globuli rossi; l’aptoglobina è assente o bassa (invece nell’emolisi extravascolare l’aptoglobina è lievemente ridotta); avremo emoglobina nelle urine, cioè emoglobinuria (presenza di emoglobina libera nelle urine) da non confondere con l’ematuria (presenza di globuli rossi nelle urine); avremo emosiderinuria, (condizione assente invece nell’emolisi extravascolare); spesso l’emolisi intravascolare presenta un test di Coombs negativo, cioè gli anticorpi contro i globuli rossi sono negativi (mentre spesso invece sono positivi gli anticorpi contro i globuli rossi nell’emolisi extravascolare) e ovviamente LDH e la bilirubina saranno aumentate in ambedue le condizioni. · Allora parlando di anemie emolitiche dobbiamo considerare le anemie da difetti intrinseci ai globuli rossi e da difetti estrinseci ai globuli rossi. I difetti intrinseci sono tutti ereditari tranne che in una condizione, cioè l’emoglobinuria parossistica notturna: questa è l’unica condizione acquisita di difetto di membrana che può provocare un’anemia emolitica da distruzione dei globuli rossi. Tutte le altre condizioni sono ereditarie e dipendono dalla fragilità del globulo rosso, fondamentalmente da difetti di membrana, ad es. la carenza di una delle proteine del citoscheletro con sferocitosi, ellissocitosi per cui il globulo rosso è più soggetto ad essere distrutto dalla milza. * Ricordiamo il famoso crash test quando i globuli rossi passano dai sinusoidi della milza: se il globulo rosso non supera quel test perché ha un difetto nella sua conformazione, nella conformazione dello scheletro di membrana, allora viene distrutto. Allo stesso modo viene distrutto se ha un difetto enzimatico che lo rende più sensibile allo stress ossidativo, per cui se mancano questi enzimi fondamentali della sua funzione glicolitica è più fragile e viene distrutto. La distruzione può essere dovuta per un’alterazione nella sintesi della globina, abbiamo visto nelle talassemie l’aggregazione delle catene globiniche in eccesso e quindi questo espone i globuli rossi ad una distruzione più rapida e più intensa. Infine può esserci un’alterazione della sintesi della globina, quindi non più un’alterazione quantitativa nella produzione di globina, come nel caso della talassemia, ma un’alterazione qualitativa del tipo di emoglobina e questo è il caso classico dell’anemia falciforme. Lezione 5 Continuiamo la lezione sulle anemie e ricordiamo sempre la semplicità della diagnosi, della diagnostica almeno di primo livello delle anemie, dove distinguiamo anemie da perdita di sangue, anemie da aumentata distruzione, ambedue caratterizzate dall'aumento dei reticolociti, e anemie da ridotta produzione caratterizzate da una bassa conta dei reticolociti. Ricordiamo l'importanza dell' MCV, quindi dei reticolociti nella diagnostica delle anemie. Abbiamo visto come le anemie si possono classificare in microcitiche, normocitiche e macrocitiche. Le anemie microcitiche sono soprattutto quelle da carenza di ferro e le sindromi talassemiche, le anemie macrocitiche sono prevalentemente da carenza di B12, le anemie normocitiche si possono avere per tanti motivi comprese le anemie da flogosi cronica, che talvolta possono anche essere microcitiche. Abbiamo parlato dell'emolisi intravascolare ed extravascolare, dell'importanza della emosiderinuria, del fatto che quando c'è un'emolisi c'è un aumento di LDH, della bilirubina, un aumento dei reticolociti ed una abbassamento dell'aptoglobina, perché ovviamente l'aptoglobina si consuma legandosi all'emoglobina libera. L'emolisi intravascolare ed extravascolare: l'emolisi extravascolare è quella che avviene nel sistema reticoloendoteliale, da parte dei macrofagi che fagocitano questi globuli rossi, quella intravascolare avviene all'interno dei vasi dove vengono distrutti i globuli rossi. Abbiamo visto che quando c'è una emolisi aumentano i reticolociti e c'è un'espansione del midollo, tipica della condizione della talassemia, dove c'è un'espansione midollare che dà poi una deformità scheletrica delle ossa piatte. Anemie emolitiche Le anemie emolitiche possono essere classificate in: anemie legate ad un difetto intrinseco ai globuli rossi e anemie invece legate ad un difetto estrinseco ai globuli rossi. A) I difetti intrinseci dei globuli rossi sono tutti ereditari tranne l'emoglobinuria parossistica notturna, che è una condizione acquisita, tutti gli altri sono difetti ereditari, ed è abbastanza semplice concettualmente questo discorso perché i difetti del globulo rosso possono essere dovuti: - ad un difetto della membrana, il globulo rosso è formato soltanto da membrana, - ad un difetto in quella modesta attività enzimatica che è presente all'interno del globulo rosso, pochi enzimi che possono essere coinvolti in questa patologia, - ad alterazioni della sintesi dell'emoglobina, o perché c'è una ridotta sintesi dell'emoglobina, per cui c'è uno sbilanciamento tra le catene alfa e le catene beta, oppure c'è una sintesi di una globina patologica che subisce delle modifiche, per esempio nell'anemia falciforme che è responsabile poi dell'emolisi. Quindi concettualmente, anche se sono tante le malattie emolitiche congenite, concettualmente si possono ridurre a tre condizioni: quella condizione in cui c'è un difetto della membrana, quella condizione in cui c'è un difetto dell'attività enzimatica del globulo rosso una condizione in cui c'è una alterazione dell'emoglobina che rende più sensibile il globulo rosso all'emolisi. 1 1) Vediamo quelle condizioni di anemie emolitiche che possono essere ricondotti ad un'alterazione, ad un disordine della membrana eritrocitaria. La più importante, la più frequente è la sferocitosi ereditaria (esistono anche condizioni molto rare di ellissocitosi ereditaria, la poichilocitosi ereditaria, insomma condizioni molto rare). La condizione comunque più importante è la sferocitosi ereditaria che è caratterizzata dalla presenza di sferociti, questi sferociti derivano dal fatto che si perde un pezzo di membrana nella milza e tutto questo modifica l'aspetto morfologico del globulo rosso che non è più a forma di lente biconcava ma è a palla, questo dipende dal fatto che ci sono delle alterazioni della parete, del citoscheletro, della membrana che impediscono al globulo rosso di avere la normale conformazione a lente biconcava. Questa malattia è caratterizzata da una modesta anemia, con una storia familiare positiva ovviamente e la presenza di ittero e anemia nel periodo neonatale, e allo striscio di sangue periferico si vedono questi sferociti, ovviamente questa deve essere distinta dall'emolisi da incompatibilità materno-fetale. Usualmente, quando c'è uno stress come un'infezione da virus, o anche la gravidanza, questo rende più evidente la sintomatologia e rende quindi più facile la diagnosi, oppure per esempio la diagnosi si fa perché si trovano dei calcoli nella colecisti e questo fa sospettare appunto che questo paziente ha avuto spesso delle crisi emolitiche con aumento della bilirubina, che è una condizione che favorisce appunto i calcoli della colecisti. Nelle forme severe sono anche presenti delle ulcere nelle gambe. quindi la maggior parte dei pazienti hanno una condizione cronica di emolisi e questo perchè questi sferociti che sono poco deformabili vengono intrappolati nelle milza, e abbiamo visto come nella milza questi globuli rossi vengono sottoposti ad uno stress: se sono ben deformabili passano i sinusoidi delle milza, se non sono ben deformabili vengono distrutti. Siccome nella sferocitosi ereditaria una delle caratteristiche è la perdita della deformabilità in questo caso aumenta la distruzione di questi elementi e quindi la condizione di anemizzazione. Inoltre, visto che la milza viene sottoposta a tutto questo lavoro chiaramente si ingrandisce e quindi un reperto che noi troviamo spesso in questi pazienti è una splenomegalia. La patogenesi è dunque dovuta ad un difetto del citoscheletro, di una della proteine che compongono il citoscheletro, può essere la spectrina, la anchirina, la banda3, ci sono diverse possibilità di difetto, che sono responsabili di questa mancata formazione corretta della membrana dell'eritrocita. Per l'ellissocitosi ereditaria fondamentalmente è lo stesso, ovviamente cambia il fenotipo. 2) Tra le anemie emolitiche che sono invece che sono legate ai difetti enzimatici il prototipo è la carenza di glucosio-6-fosfato deidrogenasi (G6PD). Esiste anche la carenza di piruvato chinasi (PK) che è più rara, e poi altre condizioni ancora più rare. La carenza di G6PD è una malattia abbastanza eterogenea ed è causata spesso da una mutazione puntiforme del gene che produce questa proteina. E' una malattia legata al sesso perché viene ereditata col cromosoma X e quindi è una condizione molto rara nelle donne, perché le donne per essere affette da questa malattia dovrebbero essere omozigoti. Tuttavia anche la forma eterozigote può avere qualche manifestazione clinica; chiaramente nelle donne è molto meno frequente l'espressione di questa malattia perché c'è l'altro X che compensa la mancata produzione di G6PD da parte di X; nell'uomo questo non è possibile. La carenza di G6PD può determinare delle caratteristiche di malattia sia acuta che cronica, ma nella maggior parte dei casi i pazienti sono assolutamente asintomatici, solo che hanno un rischio di sviluppare delle anemie emolitiche e questo avviene a causa di stress ossidativo. Una condizione che può causare lo stress ossidativo è l'ingestione di fave (o a volte anche la semplice esposizione!), ma anche le infezioni, alcuni farmaci e soprattutto appunto le fave per cui questa malattia prende il nome di favismo perché una componente delle fave è in grado di produrre dei 2 radicali di ossigeno che non vengono smaltiti dai pazienti che hanno un difetto di G6PD. Figura: in questa figura si vede la G6PD, un enzima chiave per poter ricostituire il NADPH e per poter ricostituire il glutatione che è quello che è in grado di rispondere allo stress ossidativo; quindi il NADPH prodotto da G6PD è richiesto per la rigenerazione del glutatione, che è quello che protegge la cellula dallo stress ossidativo. Se manca la G6PD manca questo sistema di protezione dallo stress ossidativo. Chiaramente siccome il globulo rosso non ha nucleo una volta consumato quel poco di G6PD che è contenuto nel globulo rosso, non se ne può formare altro e quindi questo rende la cellula sensibile allo stress ossidativo, per questo chi è carente di G6PD ha l'emolisi ma non ha un difetto in altri organi, perché questa carenza si ha non soltanto nel globulo rosso ma in tutte le cellule solo che il globulo rosso non ne può formare altro, mentre tutte le altre cellule possono formare G6PD e quindi sono resistenti allo stress ossidativo. In questa diapositiva si vede anche un altro enzima molto importante, la PK, che interviene nel metabolismo del glucosio, che voi sapete è l'unico metabolismo che il globulo rosso possiede: la mancanza di PK determina un blocco in questa via di utilizzazione del glucosio. Esistono diverse varianti della carenza di G6PD: esiste la variante africana, la variante mediterranea e poi di recente sono state scoperte nuove varianti in Cina. L'altra possibilità è quindi la carenza di PK. La PK grossomodo dà la stessa condizione di anemia e di ittero, ma la cosa interessante della PK, la cui patogenesi dipende dal fatto che viene limitata la produzione di ATP da parte della glicolisi, è che aumenta il 2,3-difosfoglicerato, questo comporta una ridotta affinità dell'emoglobina per l'ossigeno e quindi arriva più ossigeno ai tessuti: in realtà quindi i sintomi della carenza di PK sono minori rispetto al grado di anemia, perché c'è maggiore cessione di ossigeno ai tessuti e questo dipende appunto dal fatto che mancando la PK c'è un accumulo di 2,3difosfoglicerato che è quello che permette una maggiore cessione di ossigeno ai tessuti. Per cui questa condizione anemica, che dipende dal fatto che non si può produrre ATP per il fatto che questa via metabolica è alterata, tuttavia è una condizione che in qualche modo si compensa proprio per l’accumulo di 2,3 bifosfoglicerato che permette un maggiore rilascio di ossigeno. La clinica in pratica è la stessa della sferocitosi per esempio, perché è sempre una anemia emolitica e quindi c'è un aumento della bilirubina presenza di calcoli nella colecisti e chiaramente poi bisogna fare delle indagini specifiche per andare a vedere questa carenza di PK. Il favismo è una patologia abbastanza frequente, la carenza di PK è piuttosto rara. 3) Adesso passiamo alle anemie emolitiche in cui c'è un'alterazione dell'emoglobina. Una condizione che dalle nostre parti è molto frequente è l'anemia falciforme. Abbiamo già visto la scorsa lezione le alterazioni quantitative della globina che sono le talassemie, dove appunto c'è un difetto di produzione di una catena globinica quindi uno sbilanciamento tra α e β catene e la conseguenza è l'emolisi. Adesso vediamo un'altra anemia in cui c'è una alterazione della globina, ma non è un'alterazione quantitativa ma è un'alterazione qualitativa, cioè c'è una mutazione di una singola base che provoca una modifica di un aminoacido. Quando l'ossigeno è basso questa emoglobina diventa insolubile, tende quindi a precipitare, a formare dei polimeri e questo porta a delle modifiche anche della membrana eritrocitaria che viene definito a forma di falce e questa modifica, questo irrigidimento della membrana eritrocitaria porta ad una vasocclusione dei piccoli vasi. Tutta qui è racchiusa la patogenesi della sintomatologia dell'anemia falciforme, quindi questa singola alterazione che provoca l'anemia falciforme rende questa emoglobina instabile per cui si polimerizza, a differenza delle varie molecole di emoglobina, in presenza di bassa concentrazione di ossigeno e determina un'alterazione della forma e della rigidità della parete per cui si formano questi globuli rossi anomali. Quindi la deossigenazione degli eritrociti che hanno questa alterazione porta ad una polimerizzazione 3 dell'emoglobina intracellulare, il globulo rosso perde deformabilità e acquista modifiche della morfologia centrale. Quindi questi pazienti se sono ben ossigenati hanno questa morfologia (figura), i globuli rossi hanno una morfologia normale a lente biconcava, in condizione di deossigenazione, quindi in condizioni di ipossia si modifica la forma di questi eritrociti che assumono un aspetto vagamente a falce. Questa anemia falciforme è molto importante perché non dà soltanto un problema agli ematologi, anzi forse gli ematologi sono probabilmente tra quelli che si devono preoccupare di meno per i pazienti falcemici, perché come dicevamo prima la patogenesi, il fatto che si formano questi polimeri, il fatto che l'emoglobina precipita, il fatto che si formano queste modifiche a falce determinano una vasocclusione, quindi la sintomatologia del pazienti con la falcemia è quella di microinfarti nei vari organi dove questo succede e quindi ci possono essere delle condizioni acute quindi: • trombosi cerebrali, • trombosi dell'apparato respiratorio, la cosiddetta Acute Chest Syndrome, • trombosi a livello del fegato, • a livello del rene che provocherà ematuria, • a livello delle estremità, la cosiddetta Amput Syndrome e queste sono soltanto le condizioni acute, perché vi sono anche condizioni croniche: • problemi oculari; • ipertensione polmonare legata a questi continui e frequenti infarti venocclusivi a livello polmonare; • un'insufficienza cardiaca congestizia; • l'atrofia splenica, la milza è il primo organo che subisce questo effetto di vasocclusione, e quindi col passare del tempo si occludono tanti vasi per cui si dice che il pzt si autosplenectomizza , la milza va in fibrosi proprio per vasocclusione dei vasi splenici. Questa è forse l'unica malattia emolitica in cui non troverete mai la milza ingrossata, ma anzi spesso in questi pazienti c'è una milza molto piccola perché la milza va in atrofia, si dice che il pazienti si autosplenectomizza, perché la splenectomia è una delle terapie delle anemie emolitiche congenite, in questo caso non c'è motivo perché il paziente si splenectomizza da se stesso in quanto le continue vasocclusioni determinano l'atrofia; ci può essere una necrosi asettica dell'anca; spesso ci sono le ulcere perimalleolari, sempre legati alle microcclusioni, le microtrombosi che rendono difficile poi la guarigione di piccoli traumi, di piccole abrasioni a livello perimalleolare. In un paziente che ha questa condizione, fino a che il sangue è ben ossigenato non succede niente, quando il sangue è deossigenato, quando il sangue è in ipossia spuntano fuori questi blebs, queste emoglobine si polimerizzano, e poi una volta che l'emoglobina passa da un ambiente di nuovo ben ossigenato, come il polmone, si depolimerizza nuovamente. Figura: vediamo rappresentato tutto quello che può succedere in condizioni patologiche; la condizione di maggiore ipossia si ha a livello della milza, per cui spesso a livello della milza si formano questi microtrombi e la milza va in atrofia, però non succede nulla nel midollo osseo, non succede nulla nel cervello, non succede nulla nei polmoni. A volte invece si hanno delle trombosi a livello del midollo osseo e questo è responsabile di crisi dolorose: i pazienti con anemia falciforme vanno a finire al pronto soccorso per crisi dolorose molto intense che sono anche resistenti agli oppiacei, proprio perché si formano queste microtrombosi a livello del midollo osseo e danno un intenso dolore. 4 Quindi non dobbiamo aspettarci che la clinica del paziente falcemico sia legata all’anemia, ma la clinica dell'anemia falciforme è legata alle crisi dolorose e non sono rari i casi di pazienti che sono diventati "drogati" da farmaci proprio per superare queste crisi dolorose, però la crisi dolorosa con l'opportuna terapia passa: l'opportuna terapia significa ossigenoterapia, una buona idratazione e una buona alcalinizzazione, quindi se al pronto soccorso capita di dover affrontare una crisi dolorosa di un paziente con anemia falciforme quello che dovete fare, sapendo che questo dipende dalla condizione di ipossia, per prima cosa è dargli l'ossigeno, la seconda cosa da fare è fare una buona idratazione, per evitare che ci siano delle condizioni di disidratazione quindi di ispissatio sanguinis, la terza cosa da fare è un'alcalinizzazione che favorisce la minore precipitazione di queste sostanze, perché l'altra condizione che determina la precipitazione dell'emoglobina è la condizione di acidosi; in più bisogna dare l'antibiotico perché nella sindrome toracica acuta questo evento che porta alla vasocclusione avviene nel polmone. Quando avviene nel polmone? Avviene quando c'è un'infezione, quando nel parenchima polmonare c'è un'infezione il parenchima polmonare subisce l'effetto dell'infezione, quindi non è più un parenchima ben areato, ma essendoci tutte le cellule della flogosi quella zona non è più ben areata, è una zona di ipossia, si viene a creare una condizione ipossica che facilita la precipitazione dell'emoglobina e di conseguenza poi si ha l'anemia falciforme. Quindi tutte queste sindromi polmonari dipendono sempre dal fatto che una infezione ha provocato una trasformazione del parenchima polmonare come se fosse parenchima splenico, in termini di ipossia: quindi un presidio estremamente importante nella terapia di questi pazienti è la terapia antibiotica. E' quindi importante conoscere l'anemia falciforme, non per fare gli ematologi ma perché poi qualunque specialista può essere poi coinvolto in un caso con paziente con anemia falciforme, vedete come tutto dipende da dove si formano le vasocclusioni: i parenchimi più colpiti sono la milza, i polmoni e il midollo osseo ma può essere colpito qualunque parenchima compreso il cervello, oppure la necrosi asettica del femore, quindi anche chi fa il neurologo, chi fa l'ortopedico può venire a contatto con un paziente di questo tipo. B) Vediamo invece adesso le anemie emolitiche da difetti estrinseci ai globuli rossi, quindi in questo caso il responsabile non è il globulo rosso che è alterato, con l'unica eccezione dell'emoglobinuria parossistica notturna. Allora in questo caso le condizioni che possono determinare la distruzione dei globuli rossi sono: ovviamente gli anticorpi, e ci sono gli anticorpi caldi, gli anticorpi freddi e le cosiddette emolisine fredde che sono estremamente rare; oppure vi possono essere dei traumi, soprattutto in passato, quando si utilizzavano le valvole cardiache piuttosto primitive, queste rompevano i globuli rossi; a parte questa condizione che è ormai superata, le condizioni più importanti di anemie emolitiche da trauma sono quelle condizioni a cui abbiamo accennato l'altra volta in cui c'è una cosiddetta anemia emolitica microangiopatica, cioè si formano delle ostruzioni nei piccoli vasi, soprattutto si formano dei tralci di fibrina, e passando questi globuli rossi attraverso questi piccoli vasi dove ci sono questi tralci di fibrina vengono frammentati, questa è una condizione che noi riscontriamo nella coagulazione intravascolare disseminata, nella porpora trombotica trombocitopenica e nella sindrome emoliticouremica, che vedremo successivamente. 1) Quindi le condizioni più frequenti di emolisi da cause esterne ai globuli rossi, a parte le emolisi meccaniche, le condizioni più frequenti sono le cosiddette anemie immunoemolitiche, dove si formano degli anticorpi contro i globuli rossi. Queste anemie immunoemolitiche sono molto raramente idiopatiche, molto più frequentemente sono secondarie a degli altri disordini soprattutto malattie linfoproliferative : alcune malattie linfoproliferative favoriscono la produzione di autoanticorpi contro i 5 globuli rossi, la più frequente è la leucemia linfatica cronica: circa il 30% dei pazienti hanno anticorpi contro i globuli rossi. Ma ci sono anche alcuni farmaci che possono favorire lo sviluppo da autoanticorpi contro i globuli rossi e in genere questi sono anticorpi caldi, significa che reagiscono contro i globuli rossi a 37°C; meno frequenti sono le agglutinine fredde che sono appunto quelle che reagiscono contro i globuli rossi ad una temperatura più bassa. Anche qui però queste condizioni si vengono a creare a seguito di altre patologie che possono essere infettive o anche qui secondarie a processi linfoproliferativi. Quindi l'anemia emolitica autoimmune è una anemia che può essere anche indotta da alcuni farmaci, c'è una frammentazione dei globuli rossi, c'è un ipersplenismo e può essere secondaria a diverse condizioni: esiste il tipo anticorpi caldi, anticorpi freddi ma questo è un dato più laboratoristico, l a maggior parte sono secondarie: le prime a malattie linfoproliferative le altre a malattie infettive. secondarie In generale l'anemia emolitica autoimmune da anticorpi caldi ha un esordio abbastanza insidioso e il paziente lo riconosciamo perché è un paziente subitterico, ci possono essere anche delle condizioni di distruzione più rapida e in questo caso c'è proprio una emoglobinuria, in genere però l'ittero è abbastanza modesto: spesso questi pazienti sono splenomegalici però la splenomegalia, ricordiamo, è anche una condizione presente in molte malattie linfoproliferative, e non sappiamo quanto questa splenomegalia sia legata alla anemia emolitica autoimmune, quanto sia legata alla malattia linfoproliferativa, perché spesso le malattie emolitiche autoimmuni non sono altro che un epifenomeno di malattie linfoproliferative. Esiste anche la sindrome di Evans dove appunto c'è contemporaneamente un'anemia emolitica autoimmune e una piastrinopenia autoimmune e poi tutta una serie di malattie autoimmuni, come per esempio il lupus si possono accompagnare a questa condizione, ma per esempio anche l'artrite reumatoide, la sindrome di Sjogren ecc. ♥Però le condizioni più frequenti sono le malattie ipoproliferative, più spesso la leucemia linfatica cronica: questo non perché il clone linfocitario che determina la leucemia linfatica cronica produce gli anticorpi, non sono le cellule neoplastiche che producono gli anticorpi, ma è perché c'è una disregolazione dei linfociti regolatori, cioè manca una sottopopolazione di linfociti che servono a regolamentare i B linfociti e questo permette lo sviluppo di cloni che hanno un'attività autoanticorpale; le infezioni virali possono essere cause di questa situazione e ci sono anche alcuni carcinomi. ♥L'anemia emolitica da anticorpi freddi in pratica è la stessa cosa solo che c'è una maggiore acrocianosi, una condizione di vasocostrizione che favorisce un colorito bluastro della cute: anche qui l'anemia emolitica si associa a malattie linfoproliferative. 2) anemie EMOLITICHE MICROANGIOPATICHE · Una delle condizioni di anemia emolitica microangiopatica è la cosiddetta sindrome uremicoemolitica. Abbiamo visto che le anemie emolitiche microangiopatiche possono essere legate alla coagulazione intravascolare disseminata, alla porpora trombotica trombocitopenica e alla sindrome emolitico-uremica. Questa sindrome emolitico-uremica è la controparte infantile della porpora trombotica trombocitopenica e colpisce appunto i bambini, caratterizzata dalla frammentazione intravascolare dei globuli rossi, da basso numero di piastrine che vengono appunto consumate. Tutto questo meccanismo dipende dal fatto che si vengono a creare degli aggregati piastrinici e quindi si consumano queste 6 piastrine; quando questo avviene nell'ambito dei vasi renali c'è un'insufficienza renale e questo dipende da infezioni di alcuni batteri in particolare l'Escherichia Coli O157 e la Shigella Dissenteriae; negli adulti ci possono essere alcuni farmaci, in particolare la mitomicina c e alcune neoplasie, in particolare neoplasie dello stomaco che possono indurre questa condizione; nei bambini è invece legata alle infezioni di questi batteri: queste infezioni non fanno altro che determinare un danno dell'endotelio dei vasi renali, da questo endotelio dei vasi renali si libera il fattore di Von Willebrand il quale è in grado, quando non è ancora attivato, quando ancora è immaturo, di far aggregare le piastrine per cui c'è una abnorme aggregazione piastrinica a livello dei vasi renali che determina basso numero si piastrine, insufficienza renale e la frammentazione dei globuli rossi che passano attraverso questi microtrombi formati non solo dalle piastrine ma anche da fibrina e quindi c'è una condizione di anemia emolitica microangiopatica associata ad un'insufficienza renale: ecco perchè si chiama sindrome uremicoemolitica, perché c'è l'insufficienza renale e c'è l'emolisi. E questa è la controparte pediatrica di una sindrome ancora più grave, la porpora trombotica trombocitopenia che è invece presente negli adulti. · Nella porpora trombotica trombocitopenica il meccanismo è lo stesso, la causa è leggermente differente perché il difetto è in una proteasi, ma fondamentalmente si vengono a formare questi aggregati di piastrine che però non sono confinati al distretto renale ma sono un po’ dappertutto e in particolare sono a carico del sistema nervoso centrale, per cui questi pazienti hanno una grave sindrome neurologica. Quindi nella porpora trombotica trombocitopenica il paziente avrà questa pentade sintomatologica: • febbre • anemia • basse piastrine • disturbi neurologici • anomalie renali. Nella porpora trombotica trombocitopenica c'è sempre una aggregazione piastrinica ma questo dipende dal fatto che manca un enzima chiamato Adamts-13 che serve a scindere il fattore di Von Willebrand: il fattore di Von Willebrand quando viene liberato dalle cellule endoteliali è un fattore molto lungo ed esiste una proteasi che serve a scindere questi pezzi di Von Willebrand che così può svolgere la sua funzione fisiologica; se invece il Von Willebrand non viene scisso aggrega tante piastrine e favorisce la formazione di trombi e appunto nei pazienti che per un motivo congenito o acquisito hanno un difetto di questa proteasi c'è la formazione di questi lunghi "multimeri" di fattore di Von Willebrand che aggregano le piastrine e determinano tutto questo meccanismo di microtrombosi generalizzata, infatti la malattia si chiama porpora → perché questi poi alla fine sanguinano, trombotica → perché il meccanismo è legato alla trombosi, alla microtrombosi, trombocitopenica → perché c'è un consumo di piastrine. Per cui il paziente avrà la febbre (che in realtà è una febbricola), l'anemia che è un'anemia emolitica microangiopatica, quindi vedremo tutti i segni dell'emolisi, vedremo quindi la bilirubina aumentata, vedremo i reticolociti aumentati, vedremo l'LDH aumentato, vedremo l'aptoglobina bassa, vedremo la piastrine basse, segni neurologici legati a questi microtrombi che saranno più non soltanto a livello renale come succedeva nella sindrome emolitico-uremica ma saranno presenti a livello cerebrale e siccome sono dei microtrombi questi pazienti non hanno delle grosse trombosi ma hanno delle microtrombosi che a volte possono determinare soltanto delle sindromi che vengono scambiate per sindromi psichiatriche, e questo è molto importante, mi è capitato di fare la diagnosi di PTT di una paziente che stava per essere trasferita dalle malattie infettive in psichiatria, perché era una pzt che era stata ricoverata in malattie infettive per febbre che ha manifestato delle alterazione neurologiche di tipo 7 psichiatrico e la stavano per trasferire in psichiatria, poi si sono accorti che era anche piastrinopenica e per caso chiesero una consulenza ematologica. E' importante questo, perché questa sindrome non diagnosticata e quindi non curata c'è una mortalità dell'80%, cioè 8 pazienti su 10 muoiono, se diagnosticata e trattata con una terapia che si chiama plasmaferesi, in pratica si cambia il plasma, in questo modo si reintegra questo Adamts 13, in quel caso la guarigione è almeno dell'80%, quindi si capisce com'è fondamentale per questi pazienti fare la diagnosi, e per fare la diagnosi bisogna conoscere la malattia, quindi è importante che voi sappiate che questa malattia, che è una malattia rara (si vedono 2 pazienti l'anno), però fatta la diagnosi si salva il paziente, per cui questa "pentade" sintomatologica deve essere imparata: febbre, anemia, basse piastrine, sintomi neurologici e segni di insufficienza renale. Quindi la PTT è una malattia rara ma devastante: c'è un’insorgenza improvvisa di febbre, disturbi neurologici che possono essere convulsioni, coma, paralisi ecc, c'è l'anemia e in alcuni casi c'è l'emoglobinuria, c'è l'ittero perché è un'anemia emolitica, c'è quest'andamento piuttosto fluttuante, e andando a fare una valutazione istologica troviamo microtrombi attraverso il letto capillare dei piccoli vasi e sono trombi formati soprattutto da depositi di fibrina e di piastrine e questo è legato alla produzione di polimeri piuttosto larghi del fattore di Von Willebrand che tendono ad aggregare le piastrine. EMOGLOBINURIA PAROSSISTICA NOTTURNA Un altra malattia rara ma molto interessante è la cosiddetta emoglobinuria parossistica notturna (EPN). L'EPN è una malattia usualmente cronica caratterizzata da una emolisi intravascolare che dipende da un difetto di questi globuli rossi che sono più suscettibili alla lisi del complemento. In genere questi pazienti non sono solo anemici ma spesso ci sono anche altre alterazioni delle piastrine e sono anche in questo caso caratterizzati da una alta incidenza di trombosi, anche se non è molto chiaro il motivo della trombosi. In questi pazienti c'è una alterazione a carico del gene PIG-A nelle cellule staminali. Questo gene in pratica serve alla produzione di una molecola che si chiama fosfatidil-inositolo, una glicoproteina che serve ad ancorare sulla membrana cellulare altre proteine, si chiama proteina di ancoraggio, quindi mancando questa proteina di ancoraggio sulla superficie della cellula mancano le altre proteine, in particolare non si possono ancorare le molecole CD 55 (DAF) e CD 59 (MIRL), molecole che servono a bloccare l'azione del complemento, quindi queste cellule rimangono esposte, non sono più protette dall'azione del complemento, pertanto quando c'è un'attivazione del complemento, che avviene prevalentemente di notte quando si abbassa il pH, queste cellule vengono attaccate dal complemento che le lisa e quindi si ha una emolisi intravascolare. Le due proteine CD55 e CD59 sono quelle che mancando rendono la cellula sensibile all'effetto del complemento e corrispondono rispettivamente al DAF (decay accelerating factor) e a questo MIRL che sta per membrane inibitor reactive lising, cioè un inibitore di membrana della lisi reattiva dal complemento, quindi queste cellule sono sensibili all'effetto litico dell'azione del complemento e quindi c'è un'emolisi intravascolare: l'emolisi intravascolare provoca appunto una emoglobinuria, si rompe il globulo rosso, si libera emoglobina e l'emoglobina passa il filtro renale, per cui questi pzt si ritrovano al mattino con le urine particolarmente cariche, color coca-cola si dice, e questo perché l'emolisi è avvenuta di notte quando si è abbassato il pH e quando c'è maggiore sensibilità all'effetto litico del complemento. In realtà l'EPN è una malattia cronica con dei picchi di emolisi e però è difficile andare a studiare il picco di emolisi, perché appunto il paziente ha l'emolisi di notte e poi va dal medico tre giorni dopo 8 perché a parte le urine color coca-cola non ha altri sintomi immediati, i sintomi li ha dopo perché diventa anemico e comincia a star male. E allora come si fa a vedere che il paziente ha avuto l'emolisi? come dicevamo l'altra volta uno dei parametri importanti è la cosiddetta emosiderinuria, cioè l'emoglobina che ha passato il filtro renale va a depositarsi sulle cellule del tubulo renale, le quali vengono dismesse quotidianamente nelle urine e possiamo andare a dosare il ferro sul sedimento urinario dove sono presenti queste cellule: se vediamo che c'è una maggiore concentrazione di ferro quello è un segnale che c'è stata una crisi emolitica che ha provocato emoglobinuria e che quindi ha determinato l'accumulo di ferro a livello delle cellule del tubulo renale. Una cosa importante è che l'EPN è associata all'anemia aplastica e su questo sono state fatte delle speculazioni, noi non abbiamo delle informazioni sicure però c'è una teoria molto interessante che è la seguente: probabilmente l'espansione di questo clone di EPN trova spazio proprio perché c'è stata la distruzione del normale midollo quindi in realtà lo sviluppo di questa malattia è una condizione che probabilmente salva la vita a questi pazienti in quanto gli permette di superare la condizione di aplasia midollare. Se c'è una condizione che determina aplasia midollare (cioè distruzione del parenchima midollare) → se il paziente che ha questa modifica delle sue emazie (ovvero emazie EPN - ed è stato dimostrato che è una condizione molto frequente, cioè probabilmente molti di noi abbiamo un piccolo clone EPN) → quando interviene una distruzione di tutto il midollo queste emazie sono più resistenti alle cause che provocano distruzione midollare ed ecco che questo piccolo clone EPN si espande e prende il posto della normale eritropoiesi, però è un clone che ha questo difetto intrinseco e quindi è più suscettibile all'azione litica del complemento e il paziente diventa anemico: ecco spiegata, secondo questa teoria la frequente associazione tra emoglobinuria parossistica notturna e l'aplasia midollare. Quindi le caratteristiche dell'EPN sono: • l'urina colorata che durante il giorno torna normale; • dolori addominali; • trombosi venose; • trombosi cerebrali (a volte). Questo perché, come dicevamo, questo difetto non è legato soltanto ad un difetto dei protettori contro il complemento, perché proprio manca questa proteina di ancoraggio che serve ad ancorare tante altre proteine, tanti altri antigeni della membrana cellulare, quindi non sono alterati solo i globuli rossi, ma sono alterati anche le piastrine, le cellule endoteliali, c'è un'alterazione globale dell'organismo per cui in molte cellule mancano alcuni antigeni e questo favorisce lo sviluppo di fatti trombotici, favorisce altre condizioni patologiche. La cosa interessante è che adesso c'è una terapia per questa malattia ed è un anticorpo monoclonale diretto contro una frazione del complemento per cui bloccando questa frazione del complemento si blocca il meccanismo dell'emolisi; è una terapia estremamente costosa, per fortuna che i pazienti sono pochi, è una patologia abbastanza rara. 9 anemia emolitica del neonato ♥ Un'altra anemia emolitica da anticorpi è la cosiddetta anemia emolitica del neonato: in questo caso non si tratta di autoanticorpi ma si tratta di anticorpi che la mamma produce nei confronti dei globuli rossi del feto e poi del neonato. E' una malattia in cui appunto c'è la distruzione dei globuli rossi da parte degli anticorpi della mamma e può essere anche molto grave perché può causare la morte intrauterina del feto. Questa condizione dipende dal fatto che ci può essere uno scambio di globuli rossi tra la mamma e il bambino attraverso la placenta e se sono globuli rossi differenti rispetto a quelli della mamma chiaramente si producono degli anticorpi, questo succede ovviamente nel caso di incompatibilità Rh, ma può succedere anche in caso di incompatibilità AB0. Questi anticorpi se sono IgG riescono a passare la placenta e reagiscono nei confronti del bambino dandogli l'emolisi, quindi possono essere anticorpi anti-D, cioè anti-Rh, ma anche anti-AB0, e anche contro qualche altro antigene minore. Usualmente c'è questa barriera tra il bambino e la madre, rappresentata dalla placenta, però ci possono essere tante condizioni che possono rompere questa barriera (un ligando incompleto, la placenta malformata ecc), e soprattutto il parto determina una commistione di sangue tra la mamma e il bambino e quindi tutte quelle condizioni che favoriscono questo passaggio dalla circolazione fetale alla circolazione materna determinano la formazione di anticorpi, se c’è incompatibilità. Però gli anticorpi che si formano all'inizio sono IgM e le IgM sono pentameriche quindi sono grandi e non riescono a passare la placenta pertanto la prima esposizione del sangue del bambino verso la mamma determina la formazione di anticorpi da parte della mamma che sono IgM e non passano la placenta, soltanto una successiva esposizione determina il richiamo anticorpale, non produce più IgM ma produrrà IgG → quindi la incompatibiltà dipende dalla dose, dal volume dell'antigene presentato, dalla capacità dell'antigene di indurre una risposta e quanto spesso l'antigene viene presentato e se c'è incompatibilità materno-fetale. Quindi la produzione di anticorpi IgG è in grado di passare la placenta, ma se sono IgM non passano la placenta: questo significa che nella prima gravidanza è estremamente infrequente che ci possa essere una malattia emolitica del bambino, ma questo succede più spesso nella seconda gravidanza. Tenete conto però del fatto che la mamma può anche essere sensibilizzata per via di un pregresso aborto: quindi a volta la prima gravidanza è stata preceduta da un aborto e in quel caso non deve essere considerata prima gravidanza per quanto riguarda la malattia emolitica del neonato, ma deve essere considerata come se fosse una seconda gravidanza. Nel bambino con emolisi c'è una eritropoiesi compensatoria, cominciano a comparire in circolo le cellule immature (eritroblasti in circolo), c'è una eritropoiesi extramidollare. La severità dipende da molte variabili ma soprattutto dalla quantità di anticorpi che vengono formati e dal tipo di anticorpi e la malattia può essere media, moderata o severa. →Nelle forme MEDIE c'è soltanto un ittero, però ci può essere una anemia subito dopo la nascita; →nelle forme MODERATE c'è un ittero più importante e l'ittero può essere grave perché si può venire a creare quello definito kernittero, quando la bilirubina supera i 20 riesce a passare la barriera ematoencefalica e determina una tossicità ai nuclei della base per cui questi bambini posso avere delle gravi sequele neurologiche, per cui sarà capitato di vedere dei neonati messi sotto la lampada che serve appunto a distruggere le molecole di bilirubina per evitare che arrivi a valori molto alti in grado di passare la barriera ematoencefalica. E quindi i bambini possono avere questo rischio di kernittero che poi può dare spasticità, disturbi mentali, difetti cognitivi pure o anche addirittura la morte per una crisi respiratoria dovuta appunto ad 10 una tossicità dei gangli della base. →I bambini con una condizione SEVERA sono pazienti che possono anche morire in utero a causa di questa grave anemia che determina l'ipossia. Quindi è una malattia che colpisce soprattutto quando c'è una incompatibilità Rh ma ci può essere anche per incompatibilità AB0, e può essere vista anche nella prima gravidanza. Accanto all'immunizzazione dei globuli rossi con lo stesso identico meccanismo ci può essere un'immunizzazione nei confronti delle piastrine e dei globuli bianchi, sono condizioni molto più rare ma esistono, e il meccanismo è assolutamente lo stesso: c’è un’incompatibiltà materno-fetale, il passaggio di questi globuli bianchi e piastrine tra il bambino e la mamma determina lo sviluppo anticorpi contro le piastrine o contro i globuli bianchi. ±Infine vediamo una "curiosità": è rappresentata la biochimica della sintesi dell'eme! esistono delle malattie che si chiamano porfirie dove c'è un’alterazione della normale sintesi dell'eme; sono malattie abbastanza rare e che oltre a determinare anemia provocano delle altre condizioni: questi sintomi dipendono dal metabolita che si accumula, il quale dipende dal punto dove c'è la il blocco metabolico. Qui è rappresentato, per esempio, in questo percorso di formazione dall'acido δ-amino-levulinico fino alla formazione dell'eme: vediamo che c'è un difetto dell'enzima e chiaramente c'è una strozzatura in questo passaggio e si accumulano tutti i metaboliti a monte di questo enzima e a seconda del metabolita che si accumula questo va a dare tossicità ad alcuni parenchimi. In particolare alcuni metaboliti danno tossicità per il fegato, alcuni per il sistema nervoso centrale, altri per il SNP, altri per la pelle, rendono la pelle particolarmente sensibile alla luce, estremamente fotosensibile. Per cui i pazienti con porfiria possono avere tutta una serie di sintomi: compromissione dei nervi periferici, del SNC, dolori addominali, solo perché c'è una tossicità a livello del SNC e periferico che dipende dall'accumulo di questi metaboliti, inoltre possono avere tossicità epatica e questa eccessiva fotosensibilità, inoltre sono pazienti anemici. Probabilmente la leggenda del lupo mannaro dipende proprio da pazienti affetti da alcune forme di porfiria, perché erano pazienti anemici, pazienti che avevano una particolare sensibilità alla luce e quindi dovevano uscire di notte quando c'era la luna piena altrimenti non ci vedevano e avevano delle alterazioni cutanee per questa fotosensibilità caratterizzate da un iperirsutismo che faceva crescere molti peli, avevano un aspetto molto irsuto, uscivano soltanto di notte e si nutrivano di sangue perché il sangue a quel tempo era l'unico rimedio per combattere l'anemizzazione e da lì la leggenda. 11 Lezione 6 Il prof si lamenta un po’ del risultato del primo test sulle anemie. 1. La sintomatologia dell’anemia dipende da: Rapidità di insorgenza dell’anemia è la risp esatta. 2. Le ulcere peri-malleolari sono tipiche dell’anemia falciforme. La patogenesi dell’anemia falciforme è legata alla formazione di microtrombi. Poiché in condizioni di ipossia i globuli rossi falcizzano, ciò crea dei microtrombi che determinano dei microinfarti. Tutto questo determina le crisi acute dolorose e le ulcere perimalleolari sempre a causa dei microtrombosi che intervengono a carico di una zona poco irrorata come le zone perimalleolari, spesso soggette a traumi che non quindi guariscono. 3. Nell’anemia da flogosi cronica la ferritina è usualmente elevata. La differenza tra l’anemia sideropenica e quella da flogosi cronica è proprio la valutazione della ferritina. In genere l’anemia da flogosi cronica è normocitica però può essere anche microcitica. Quindi essendo microcitica viene posta diagnosi differenziale con l’anemia sideropenica e la talassemia. Nell’anemia sideropenica la ferritina è bassa perché mancano i depositi, viceversa nell’anemia da flogosi cronica la ferritina è aumentata perché per la presenza di alcune citochine e soprattutto per un’elevata produzione di epicidina il ferro viene bloccato, cioè il ferro non passa in circolo, non viene assorbito dall’intestino nel sangue e non passa dai macrofagi nel sangue. Per cui il ferro si accumula nei depositi però si abbassa nel sangue, pertanto la sideremia sarà basso, come succede nell’anemia sideropenica. La sideremia è bassa in entrambe le condizioni quello che varia è al ferritina. 4. La causa più frequente di carenza di ferro è l’eccessiva perdita (70% dei casi). Nelle donne soprattutto in età fertile è legata alle perdite mestruali ma può essere dovuta alle perdite gastro-enterica prevalentemente negli uomini. Quindi se facciamo diagnosi di anemia sideropenica in un uomo, chiaramente dobbiamo andare a ricercare la perdita nel tessuto gastro-enterico; in una donna in età fertile andiamo a indagare su durata, frequenza delle mestruazioni. Meno frequentemente l’anemia sideropenica dipende da un insufficiente assorbimento, come può succedere in tutte le condizioni in cui l’intestino soffre di qualche patologia oppure c’è un abuso di inibitori di pompa o qualunque condizione impedisca la trasformazione del ferro da trivalente a bivalente e pertanto il suo assorbimento. 5. Nella carenza di ferro il parametro che si riduce più precocemente è il ferro di deposito cioè la ferritina. Successivamente inizia la riduzione del ferro circolante e infine inizia l’anemia (poiché una volta che manca il ferro il midollo non può più produrre globuli rossi, Hb). 6. Nel morbo di Cooley il paziente presenta valori di ferritina aumentati. La patogenesi della talassemia è quella di uno squilibrio delle catene, ciò provoca una formazione di aggregati insolubili che precipitaziono. Tutto questo determina la formazione di eritroblasti anomali che vengono rapidamente distrutti. In questi pazienti vengono fatte delle trasfusioni e tutto ciò determina un sovraccarico sistemico di ferro e quindi emocromatosi secondaria, un eccesso di ferro → tant’è vero che uno dei cardini del trattamento dei pazienti talassemici è proprio la terapia ferro-chelante, la somministrazione di farmaci che chelano il ferro ed evitano a questo di andarsi a depositare a livello del cuore, del fegato, delle isole pancreatiche: la causa mortis dei pazienti talassemici è proprio l’insufficienza cardiaca che dipende in parte dall’aumento della gittata cardiaca dovuta all’anemia ma in gran parte dal sovraccarico di ferro a livello cardiaco e anche a livello del fegato (per cui questi pazienti diventano cirrotici), delle isole pancreatiche (questi pazienti diventano diabetici), le ghiandole endocrine (i pazienti non crescono anche per questo motivo, si ha ipogonadismo). In definitiva è proprio l’emocromatosi secondaria più che l’anemia ad essere dannosa a lungo termine per questi pazienti. Questo per ricordare che quindi nel morbo di Cooley i valori di ferritina (che rappresenta il ferro di deposito) sono sicuramente aumentati. 7. Per il portatore di talassemia è sempre controindicata la terapia marziale? Il prof ha inserito questa domanda perché c’è un malinteso in generale tra gli ematologi e la classe medica. Come visto in precedenza il paziente talassemico usualmente ha valori di ferritina aumentati, cioè possiede una quantità di ferro più elevata della norma, allora una volta (quando questi concetti non erano ben chiari, ad es. non si faceva la determinazione della ferritina) a tutti i pazienti anemici gli si dava il ferro, questo perché l’anemia sideropenica è indubbiamente la condizione più ferquente di anemia. Ma allora gli ematologi hanno detto che il paziente talassemico di ferro ce ne ha già di suo abbastanza, quindi è inutile dare altro ferro, anzi questo è in qualche modo controindicato → tutto questo è stato interpretato in maniera malintesa considerando come se nel paziente affetto da morbo di Cooley ci fosse una controindicazione assoluta a dare il ferro!! (ecco dov’è il malinteso) Infatti capita molto spesso di vedere pazienti che sono portatori di talassemia ma che sono anche per qualche altro motivo sideropenici e ai quali il medico curante non dà il ferro manco a morire!! perché pensa che sia controindicato. In realtà quindi non c’è nessuna controindicazione, è semplicemente un discorso di valutazione dell’assetto marziale, infatti nulla vieta che un paziente talassemico per qualche altro motivo (perché ha le mestruazioni o una perdita a livello del colon) nulla vieta che il paziente possa diventare sideropenico e in quel caso deve essere somministrato il ferro. Quindi non c’è nessuna controindicazione assoluta nel somministrare il ferro ai pazienti con talassemia. È chiaro che se la causa dell’anemizzazione è la talassemia non bisogna somministarre il ferro. 8. Tutte le seguenti affermazioni sono vere tranne una, nell’anemia falciforme: c’è un rischio di trombosi cerebrale è frequente la necrosi asettica dell’anca possono presentarsi ulcere perimalleolari la splenectomia non è necessaria nella maggior parte dei pazienti, perché in realtà a causa di fenomeni di micro-ostruzione vascolare si dice che il paziente si autosplenectomizza, esiste una fibrosi splenica legata proprio ai microtrombi; la milza è un organo ipossico e il paziente va in atrofia splenica. La necrosi ischemica cronica della milza alla fine porta all’atrofia → quindi è una delle pochissime anemie emolitiche in cui non c’è bisogno di fare splenectomia perché è la malattia stessa che determina una condizione di splenectomia. 9. Nelle anemie emolitiche da difetto intraglobulare (dove c’è un’alterazione legata al globulo rosso) l’unica forma acquisita è l’emoglobinuria parossistica notturna. L’EPN è una condizione in cui viene a mancare la produzione di una proteina di ancoraggio della membrana eritrocitaria (fosfatidil-inositolo), che serve ad ancorare sulla superficie delle cellule alcune molecole, tra le più importanti vi sono le molecole che inibiscono il complemento DAF e MIRL: mancando queste molecole che proteggono la cellula dal complemento, la cellula diventa più sensibile all’azione litica mediata dal complemento. Pertanto quando si attiva il complemento per una condizione ad es. di acidosi, il complemento aggredisce queste cellule e provoca una emolisi → si tratta di una emolisi intravascolare per cui si rompe il globulo rosso, si libera l’emoglobina la quale passa il filtro glomerulare e quindi c’è emoglobinuria Malattie Linfoproliferative In ematologia questo costituisce un grosso capitolo, sono malattie neoplastiche del sistema linfatico e la manifestazione clinica più evidente di queste malattie sono le linfoadenopatie, cioè l’ingrossamento dei linfonodi. Però l’ingrossamento dei linfonodi non è dovuto soltanto alle malattie linfoproliferative, ma qualunque stimolazione infiammatoria come sappiamo determina l’ingrossamento dei linfonodi. Quindi la diagnosi di malattie linfoproliferative va fatta sempre attraverso la biopsia di un linfonodo, però è importante che il clinico, il medico sappia quando andare a fare la biopsia..non è che possiamo biopsare tutte le linfoadeniti!! Quindi occorre un minimo di discernimento, quando si presenta un paziente con una linfadenopatia dobbiamo saper distinguere se questa è una linfadenopatia che ha il sospetto di essere una malattia linfoproliferativa o meno. Semeiologia delle linfoadenopatie Ci sono pertanto delle caratteristiche semeiologiche dei linfonodi che ci distinguono le 4 condizioni che possono determinare linfoadenopatie (ingrossamento dei linfonodi): 1. Linfoadenite acuta, sono abbastanza facili da diagnosticare perché i linfonodi sono dolenti e talvolta ricoperti da cute arrossata (esattamente come una tonsillite acuta). 2. Linfoadeniti croniche, cioè le pregresse affezioni infiammatorie, dove i linfonodi sono di consistenza aumentata, elastici, mobili e non dolenti. 3. Sono difficili da distinguere dai linfonodi che si ingrossano a causa delle malattie linfoproliferative, ma in questo caso i linfonodi sono un po’ più duri, la caratteristica più importante è che spesso sono agglomerati; c’è una lieve riduzione della mobilità, non o poco dolenti. 4. Metastasi da tumori solidi, i linfonodi sono duri-lignei, fissi sui piani profondi, non dolenti; talvolta la cute soprastante assume colorito rosso-bluastro e può ulcerarsi. Questo usualmente non si ritrova nei linfomi anche se qualche caso di linfoma di Hodgkin può dare questo tipo di consistenza. Un altro aspetto molto importante che serve ad orientarci se dobbiamo mandare il paziente dal chirurgo a fare la biopsia linfonodale o meno è la sede, la localizzazione anatomica. Localizzazione anatomica - Sopraclaveari, cercare sempre la neoplasia, sono da considerare neoplastici fino a quando non viene dimostrata l”innocenza”. Perché è molto difficile che il linfonodo di una linfoadenite sia un linfonodo sopraclaveare: tipica localizzazione sopraclaveare di linfonodo aumentato è data dal linfoma di Hodgkin, la cui tipica presentazione è il linfonodo sopraclaveare sx. - Epitrocleari, quasi sempre espressione di malattia linfoma tosa ma ovviamente non nel caso di una linfadenite acuta; cioè i linfonodi epitrocleari con caratteristiche non dolenti, di consistenza duro-elastica è più probabile che sia di natura neoplastica e non infiammatoria. Se invece è molto dolente, con cute arrossata e c’è una ferita nella mano o nel braccio, quella è una linfadenite reattiva. Tipica è la malattia da graffio di gatto, dove a causa del graffio del gatto ci sono alcuni batteri che possono determinare questa malattia (può dare frequentemente un linfonodo epitrocleare). -Retronucali, sono quasi sempre benigni. -Mediastinici, bisogna andare a considerare a quale parte del mediastino appartengono, se anteriore, media o posteriore perché chiaramente drenano strutture diverse e quindi possono configurare malattie diverse. -Ilari. Ricordiamo che nel torace non ci sono i linfonodi mediastinici ma anche i linfonodi ilari, dell’ilo polmonare e in questo caso i linfonodi ilari possono essere aumentati sia per malattie neoplastiche (neoplasie polmonari e linfomi), sia anche per la sarcoidosi. Anzi è più frequente che i linfonodi ilari vengano interessati dalla sarcoidosi piuttosto che da un linfoma. -Inguinali, sono spesso sedi di flogosi aspecifiche, perché drenano non solo gli arti inferiori ma anche l’apparato uro-genitale. Segni clinici Oltre alla localizzazione anatomica dobbiamo fare una corretta anamnesi del paziente per vedere se ha un linfoma o una linfoadenite. Alcuni sintomi ci possono orientare, in particolare i sintomi legati alle malattie linfoproliferative in generale sono: 1. Dimagrimento 2. Febbre 3. Sudorazioni notturne profuse Questa è la tipica triade sintomatologica delle malattie linfoproliferative, “ sintomi B” a queste si può associare un altro sintomo: • Il prurito sintomo che può anche precedere di molto tempo la diagnosi del linfoma di Hodgkin, deve essere un prurito non associato a lesioni cutanee (prurito sine materia), a parte le lesioni da grattamento secondarie al prurito. In un paziente giovane ricordiamo quindi la possibilità che il prurito può essere un sintomo molto importante per la diagnosi di Linfoma di Hodgkin (per ricordare meglio andate a vedere il film di Nanni Moretti “Caro Diario” – ultimo episodio Medici) • Dolore dopo ingestione di alcool, un sintomo molto raro, però quasi patognomonico di Linfoma di Hodgkin. Importante è visitare il paziente: • Splenomegalia, non è solo sintomo di malattia linfoproliferativa, ma anche le infezioni danno splenomegalia (mononucleosi..) • Rx torace, grossa massa mediastinica linfonodale – a quel punto il sospetto di processo linfoproliferativo è molto forte, poi per fare la diagnosi occorrerà fare la biopsia • Esami di laboratorio, alterazioni all’emocromo Il punto fondamentale di fronte a un paziente con una linfadenopatia è decidere se fare la biopsia linfonodale o meno, anche qui bisogna fare qualche osservazione che può essere utile nella gestione di questi pazienti. Quando fare la biopsia? • La diagnosi di processo linfoproliferativo è sempre una diagnosi istologica, quindi prelevare il linfonodo e analizzarlo. Mai fare una FNA, cioè l’agoaspirato dei linfonodi, questa è una pratica assolutamente sbagliata perché per fare una corretta diagnosi di un processo linfoproliferativo il patologo deve esaminare l’intero linfonodo per vedere come viene sovvertita la struttura, l’architettura del linfonodo. • In molti processi linfoproliferativi c’è un andamento a fisarmonica, il “Wax and Wane”, cioè questi linfonodi non crescono progressivamente e basta, ma a volte possono esserci delle riduzioni anche spontanee dei linfonodi; addirittura esiste anche la remissione spontanea dei linfomi, soprattutto come vedremo nei linfomi a basso grado di malignità. In questi pazienti non è infrequente osservare una regressione spontanea della linfadenopatia, come se i linfomi (anche se non guariscono) vanno in remissione spontaneamente (per es il paziente con un’infezione produce una grossa quantità di INF endogeno il quale riduce in maniera significativa il processo linfoproliferativo). • È preferibile evitare i linfonodi inguinali, poiché come dicevamo sono spesso sede di una flogosi aspecifica e la sovrapposizione del processo flogistico può determinare difficoltà interpretative del processo neoplastico. • Una vigile osservazione per qualche settimana non è irragionevole, è molto raro di trovarsi di fronte a pazienti che non possono aspettare qualche settimana per la valutazione diagnostica precisa. • È sempre possibile ripetere una biopsia. Se facciamo la biopsia linfonodale e l’anatomo patologo ci dice che è una linfoadenite, non è detto che quel paziente non abbia anche un linfoma! A volte si sbaglia a prelevare il linfonodo o si prende un linfonodo troppo satellite rispetto alla neoplasia oppure non sempre la neoplasia si manifesta, infatti è frequente che alcuni linfomi di Hodgkin (in particolare i linfomi di Hodgkin a prevalenza linfocitaria) vengono diagnosticati anche dopo la seconda biopsia linfonodale perché a volte simulano delle condizioni di linfoadenite. • Attenti alle malattie virali, se c’è una malattia virale in corso (ad es una rosolia) non va fatta la biopsia perché potremmo indurre il patologo in errore. La malattia da graffio di gatto e la mononucleosi infettiva sono condizioni che determinano linfoadenopatie, ma li studieremo in malattie infettive. LINFOMI I linfomi noi li distinguiamo in: 1. Linfomi di Hodgkin 2. Linfomi non di Hodgkin I linfomi sono malattie neoplastiche di cellule native del tessuto linfoide, sono quindi malattie che nascono nel tessuto linfoide, nascono nel linfonodo. Linfoma di Hodgkin Il linfoma di Hodgkin è una malattia ben definita e particolare: se prendiamo un linfonodo di un soggetto con linfoma di Hodgkin, la cosa peculiare è che ci sono poche cellule neoplastiche ovvero le cellule Reed-Sternberg e tante cellule reattive che sono linfociti-istiociti-eosinofili-plasmacellulefibroblasti. Quindi contrariamente a tutte le altre neoplasie in questo caso vi sono poche cellule neoplastiche (< 5%), la gran parte sono cellule normali, questa è una caratteristica unica tra le neoplasie. • Ha un’epidemiologia bimodale, cioè colpisce gli adolescenti e i giovani adulti tra i 15-34 anni e questo è il primo picco; il secondo picco invece dopo i 50 anni. Esistono evidenze epidemiologiche di associazione tra l’infezione dal virus di Epstein Barr e lo sviluppo dell’Hodgkin: • Una pregressa infezione da EBV è dimostrabile nel 50% dei casi di Hodgkin • È dimostrato che i soggetti con una storia di mononucleosi hanno un rischio da 3 a 5 volte maggiore di sviluppare il linfoma Hodgkin • Con metodiche di biologia molecolare nel 70% dei casi di Hodgkin sono state dimostrate delle sequenze del DNA di EBV dentro le cellule di Reed-Sternberg. Quindi è ipotizzabile che vi sia nella patogenesi del linfoma di Hodgkin un’attività dell’EBV o di altri virus (per es il morbillo), questi probabilmente stimolano il meccanismo, una pathway molecolare che rende immortalizzate queste cellule. Intanto è stato dimostrato che queste cellule Reed Sternberg derivano da linfociti B. Fino a qualche tempo fa infatti non si sapeva neppure a quale stipite cellulare in realtà appartenessero, soltanto di recente studiando su queste cellule il riarrangiamento VDJ è stato dimostrato che appartengono a cloni B linfocitari. Perché questi B linfociti così riarrangiati in maniera non funzionale non vanno incontro ad apoptosi? Voi sapete che nel centro germinativo del linfonodo, quando c’è un linfocita che subisce un riarrangiamento e questo riarrangiamento non è funzionale alla attività svolta contro uno specifico antigene, questi linfociti vano incontro ad apoptosi. Nel caso specifico i linfociti con questo tipo di rirrangiamento non funzionale, non muoiono e qui probabilmente c’è un ruolo del virus EBV che immortalizza queste cellule. (slide) Questo è un quadro citologico di un’apposizione sul vetrino di un linfonodo di un paziente affetto da Hodgkin, questa cellula nucleata è la cellula Reed-Sternberg (RS), tutte le altre sono le cellule normali attorno alla cellula RS → prima si pensava che fosse una reazione dell’organismo alla malattia neoplastica, in realtà è dimostrato che le cellule RS traggono nutrimento da tutte queste cellule accessorie, in qualche modo la cellula di Sternberg riesce ad assoggettare a sé tutte le cellule normali per cui provoca una sorta di infiammazione che si esplica con l’arrivo nel linfonodo di tutte le cellule infiammatorie. Classificazione La classificazione istologica si basa sulle cellule Reed-Sternberg e le sue varianti per cui avremo: 1. Linfoma di Hodgkin a prevalenza linfocitaria, nodulare. 2. Linfoma di Hodgkin classico Un tempo la classificazione prevedeva soltanto il linfoma di Hodgkin classico che a seconda della quantità di cellule Reed-Sternberg e di cellule mononucleate distingue: • Linfoma di Hodgkin ricco di linfociti, ovvero poche cellule RS e molti linfociti • Linfoma di Hodgkin con sclerosi nodulare, dove la caratteristica istologica sono questi tralci fibrosi che tagliano il linfonodo. • Linfoma di Hodgkin a cellularità mista, dove è particolarmente florida la componente reattiva e c’è un polimorfismo cellulare, ci sono eosinofili, istiociti, linfociti, plasmacellule. • Linfoma di Hodgkin a deplezione linfocitaria dove c’è un eccesso di cellule RS e percentualmente si riducono i linfociti. Queste distinzioni riguardano il cosiddetto linfoma di Hodgkin classico; accanto a questo esiste il linfoma di Hodgkin a predominanza linfocitaria nodulare, il quale probabilmente è un linfoma di Hodgkin leggermente diverso, un po’ più spostato verso la maturazione dell’ontogenesi linfocitaria, infatti l’immunofenotipo del linfoma di Hodgkin classico è differente rispetto al linfoma di Hodgkin a prevalenza linfocitaria nodulare. Immunofenotipo 1. Il Linfoma di Hodgkin classico, il più frequente 90%, sono caratterizzati dalla positività di questi due antigeni CD30, CD15. 2. Il linfoma a predominanza linfocitaria nodulare, spesso è CD30 e CD15 negativo ma ha positività per il CD20, questo è un marcatore B-linfocitario un po’ più maturo. Al giorno d’oggi quasi tutti gli anatomo-patologi che fanno diagnosi di linfoma di Hodgkin vanno a cercare la conferma di questa diagnostica, facendo tali reazioni di immuno-istochimica che evidenziano la positività per il CD30 e il CD15. L’aspetto da ricordare è che il Linfoma di Hodgkin classico è sempre CD30 e CD 15 positivo. Stadiazione Quando facciamo diagnosi di Linfoma di Hodgkin dobbiamo fare una mappa della malattia, dobbiamo fare una stadiazione e questa la facciamo secondo una classificazione di Cotswolds, distinguiamo 4 stadi: 1. STADIO I c’è l’interessamento di una singola struttura linfonodale o regione linfonodale, non singolo linfonodo. 2. STADIO II interessamento di 2 o più regioni dallo stesso lato del diaframma, si considera il corpo diviso in due dal diaframma. Quindi lo stadio II non dipende dal numero di stazioni linfonodali interessate ma dal fatto che sono interessate le stazioni da un solo lato del diaframma. 3. STADIO III interessamento di strutture o stazioni linfonodali da entrambi i lati del diaframma (sopra e sotto diaframmatiche), non dipende dal numero delle stazioni interessate. 4. STADIO IV interessamento di 1 o più sedi extralinfonodali (polmonare, osseo, epatico) Il Linfoma di Hodgkin si propaga per contiguità linfonodale, quindi non salta stazioni linfonodali, cioè un paziente con interessamento dei linfonodi latero-cervicali non può avere interessati i linfonodi para-aortici senza l’interessamento di tutta la catena interposta!! Presentazione clinica 1. Sintomi B: febbre- dimagrimento-sudorazioni notturne. Se un paziente presenta uno di questi sintomi viene classificato come B, cioè come sintomatico. 2. Sintomi A: se non ha nessuno di questi sintomi B, allora viene classificato come A (asintomatico). Il prurito come sappiamo è un sintomo molto frequente nell’Hodgkin, anzi può essere il primo sintomo ma non è considerato per definizione nei sintomi B, pertanto il paziente con prurito viene considerato A. Quindi oltre allo stadio I, II, III, IV bisogna aggiungere il suffisso A o B a seconda che il paziente sia asintomatico oppure presenta uno o più di questi sintomi B. Caratteristiche: • Una caratteristica peculiare è l’interessamento linfonodale centrale (latero-cervicali, mediastinici, para-aortici, iliaci), è difficile che il linfoma di Hodgkin dia localizzazione ai linfonodi mesenterici o epitrocleari. • 30% interessamento splenico. • 90% frequente interessamento mediastinico • Rare altre sedi In termini anatomo-patologici c’è una forte reazione infiammatoria alle cellule RS (ovvero alle cellule neoplastiche dell’Hodgkin), tutto questo si traduce in un aumento dei marcatori dell’infiammazione: • VES-fibrinogeno-calcemia-cupremia • Leucocitosi neutrofila, proprio tipica da infiammazione (non c’è una linfocitosi come ci si potrebbe aspettare, anzi questi pazienti hanno una linfopenia!) • Diminuizione della risposta cellulo-mediata Quindi molto spesso il medico, prima ancora di fare la biopsia linfonodale, ha un orientamento se il paziente ha il linfoma di Hodgkin o non-Hodgkin, proprio perché vi sono delle caratteristiche cliniche che li differenziano. Differenze cliniche Linfoma di Hodgkin: • Più spesso localizzato ad un solo linfonodo • Colpisce più frequentemente i linfonodi del gruppo assiale, cioè cervicali, mediastinici e para-aortici e si propaga in maniera ordinata per contiguità. • Linfonodi centrali, molto raramente i linfonodi mesenterici e l’anello del waldeyer • Raro coinvolgimento extranodale • Condizione infiammatoria Linfomi non Hodgkin • Localizzazione linfonodale più diffusa • Si propaga per via ematogena, può saltare alcune stazioni linfonodali • Interessa spesso i linfonodi mesenterici e l’anello del waldeyer • Frequente coinvolgimento extranodale • Non esiste la condizione infiammatoria Per cui nel linfoma di Hodgkin troveremo gli indicatori di flogosi aumentati (VES, proteina C reattiva, fibrinogeno), condizione questa che è molto meno frequente nei linfonodi non Hodgkin. Prognosi Il linfoma di Hodgkin è stato e continua ad essere uno dei cavalli di battaglia dell’ematologia e dell’oncologia, nel senso che da tempo si conosce come guarire. Il Linfoma di Hodgkin è una malattia assolutamente guaribile, ma non tutti i opazienti guarscono, esiste una quota intorno al 20-30% di pazienti che non guariscono. Allora bisogna fare una valutazione prognostica, poiché ci sono alcuni fattori importanti per prevedere se il paziente guarirà o meno, in base alla gravità → questo è importante per modulare la terapia, se abbiamo pazienti che hanno delle caratteristiche molto favorevoli li trattiamo con la terapia minima e indispensabile e sappiamo hanno delle ottime probabilità di guarire; viceversa pazienti che hanno delle caratteristiche cliniche di presentazione molto sfavorevoli, occorre che ci imbattiamo in una terapia molto più aggressiva per cercare di farlo guarire. Quindi la valutazione prognostica pre-terapia è estremamente importante e una delle valutazioni prognostiche più importanti riguarda lo stadio di malattia. Fattori prognostici stadio I-II Lo stadio della malattia: I-II sono da considerare favorevoli; III-IV non favorevoli. Ma questo è abbastanza ovvio in quanto I e II identificano malattie più limitate mentre III e IV stadio identificano malattie più estese, il carico di malattia tumorale, la quantità di cellule neoplastiche è differente. Tuttavia anche all’interno degli stadi possiamo avere dei fattori prognostici, ad es. per il I e II stadio: 1. L’età, superiore a 50 anni sfavorevole. 2. Sesso femminile più favorevole del maschile 3. Tipo istologico: es l’istotipo a prevalenza linfocitaria è più favorevole rispetto l’istotipo a deplezione linfocitaria (è abbastanza intuitivo poiché cambia il numero di cellule di Sternberg). 4. Pazienti con una Ves molto elevata hanno una prognosi più sfavorevole. 5. Nell’ambito del II stadio, se ha 4 o 5 stazioni linfonodali interessate va peggio che se ne ha solo 2. 6. Interessamento mediastinico, cioè la massa tumorale del mediastino: se i linfonodi mediastinici sono superiori ad 1/3 del diametro toracico, quindi il rapporto diametro mediastino/ torace è maggiore di 0,35 vanno peggio, questo elemento configura una prognosi sfavorevole ma anche questo è abbastanza ovvio perché se un paziente ha una grossa adenopatia mediastinica ha una prognosi peggiore, questo dipende probabilmente anche dalla fibrosi che si accompagna all’Hodgkin poiché probabilmente in questa massa tumorale i farmaci non arrivano, diventa un meccanismo di resistenza alla chemioterapia dell’Hodgkin. * Questi fattori prognostici valgono per i pazienti in stadio limitato di malattia (I e II stadio), mentre per i pazienti in stadio avanzato di malattia ci sono altri fattori prognostici che sono stati identificati una decina di anni fa (siamo nel 2010) e sono da considerare sia come estensione di malattia che come risposta dell’organismo. Fattori prognostici stadio avanzato • Albumina sierica < 40 g/dl, una bassa albumina sierica significa che la malattia ha determinato una compromissione tale per cui l’organismo non è in grado di produrre albumina. • Hb <10,5 g/dl, compromissione importante in parte legata all’anemia da flogosi cronica. • Sesso maschile • Stadio IV • Età > 45 anni • WBC > 15 x 10^9 /l (numero di globuli bianchi) • Linfociti <0,6 x 10^9/l o <8 di WBC L’aspetto curioso è che ognuno di questi parametri fa abbassare la curva di guarigione di una quantità pressocchè costante, cioè ogni fattore prognostico ha pressocchè lo stesso valore. Per cui pazienti con malattia avanzata (stadio III o IV) ma che non hanno nessuna di queste caratteristiche prognostiche viste prima (bassa albumina, bassa Hb, globuli bianchi alti etc) hanno l’80% di probabilità di guarire nonostante siano in malattia avanzata. Man mano che si aggiungono questi fattori: • 5 fattori 40% probabilità di guarigione Queste sono tutte considerazioni che vanno fatte prima di iniziare il trattamento, ma al giorno d’oggi la novità consiste nel fatto che questi pazienti li studiamo anche con nuove indagini di imaging, in particolare la PET. La Tomografia ad emisssione di positroni viene fata in questi casi non solo all’esordio della malattia ma viene fatta anche per valutare la risposta al trattamento e per convenzione si è deciso di fare la PET dopo 2 cicli di terapia: questa valutazione di PET dopo 2 cicli di chemioterapia è diventato adesso il fattore prognostico più importante (nei pazienti con malattia avanzata). Trattamento L’intento del trattamento dell’Hodgkin è sempre, in ogni stadio, di tipo curativo, cioè fare guarire il paziente. La guarigione è legata a quanto viene fatto in termini di chemioterapia e radioterapia ma negli ultimi anni si è imparato un po’ come usare questi farmaci * Il prof dice che non è necessario per noi conoscere come si tratta il linfoma di Hodgkin (interessa gli specialisti), quello che dobbiamo sapere in qualità di studenti è che abbiamo imparato una lezione dal trattamento dell’Hodgkin; infatti siccome è da tanti anni che si riesce a far guarire dal linfoma di Hodgkin (dagli anni ’70) abbiamo un follow up sufficientemente lungo per capire se è stato fatto bene o è stato fatto male. Secondo una vecchia casistica mostrata dal prof. circa la metà dei pazienti sono deceduti: • Nel contesto dei 50% morti, i deceduti per linfoma di Hodgkin sono soltanto una piccola fetta!! La stragrande maggioranza di questi pazienti è morta per altri motivi che non sono correlati all’Hodgkin, sono morti per complicazioni, in particolare per neoplasie secondarie e per le malattie cardiovascolari (i pazienti sviluppavano spesso una coronaropatia dovuta all’irradiazione a livello del mediastino, alla radioterapia). Il prof mostra anche una casistica di bambini che sono stati curati e guariti dal linfoma di Hodgkin ma che poi a distanza di 30 o più anni hanno sviluppato il tumore della mammella e questo è stato legato alla chemioterapia + radioterapia che una volta si faceva in maniera estesa a questi pazienti. Invece i pazienti trattati con la sola chemioterapia hanno una bassa incidenza di neoplasie secondarie. !! Quindi la lezione che abbiamo imparato è la seguente: è stato dimostrato che a lungo termine è più tossica la radioterapia che la chemioterapia in termini di sopravvivenza. Quindi la situazione si è ribaltata rispetto a qualche anno fa in cui invece si abbondava con la radioterapia perché si pensava che la chemio fosse più tossica! Lezione 7 Linfomi non- Hodgkin La volta scorsa abbiamo parlato del linfoma di Hodgkin, oggi parliamo dei linfomi non-Hodgkin che costituiscono una patologia di sicuro più frequente dei linfomi di Hodgkin perché contrariamente al linfoma di Hodgkin che ha una curva di incidenza di tipo bimodale, cioè con un incremento della frequenza del linfoma di Hodgkin in un picco nella fase giovanile e un secondo picco nell'età anziana, qui invece l'incidenza dei linfomi non-Hodgkin è progressivamente aumentata mano a mano che si passa dal bambino all'anziano. Quindi tipicamente è una malattia che tende a crescere negli ultimi anni e tende a crescere per diversi motivi, prima di tutto perché essendo una patologia dell'anziano mano a mano che invecchia la popolazione c'è una maggiore incidenza di questo tipo di malattia. Ma aumenta anche perché è in aumento nei pazienti immunodepressi e aumentano sicuramente le condizioni che determinano immunodepressione, sia perché si utilizzano sempre più farmaci immunosoppressori per il trattamento di patologie per esempio autoimmuni, sia perché ci sono alcune condizioni che provocano immunosoppressioni tipo il virus HIV che provoca appunto una condizione di immunodepressione che favorisce l’aumento dei linfomi. (slide) Quindi questa è una casistica inglese dove su ventimila pazienti il 90% è rappresentato da linfomi non- Hodgkin e il 10% dai linfomi di Hodgkin. Questo per rendervi l'idea di quanto sia la differenza tra l'incidenza del linfoma di Hodgkin e l'incidenza dei linfomi non-Hodgkin. Il linfoma non-Hodgkin è molto ma molto più frequente del linfoma di Hodgkin. Nell'ambito dei linfomi non-Hodgkin la stragrande maggioranza di questi pazienti hanno un linfoma che deriva dai linfociti B, mentre una piccola quota di questi linfomi deriva dai linfociti T. Questo per darvi l'ordine di grandezza di quello di cui stiamo parlando. Stiamo parlando di patologie abbastanza frequenti che sono prevalentemente derivate da neoplasie che originano dalla linea B-linfocitaria. Mentre il linfoma di Hodgkin è una patologia tipicamente dei linfonodi, cioè sono patologie che nascono comunque nei linfonodi e poi possono anche coinvolgere siti extralinfonodali. Se ricordate il IV Stadio del linfoma di Hodgkin è proprio caratterizzato dall'invasione dei tessuti extramidollari. Nel linfoma non- Hodgkin invece 2/3 dei casi originano da strutture linfonodali, ma esiste 1/3 dei pazienti con linfoma non-Hodgkin dove l'interessamento iniziale non riguarda i linfonodi ma riguarda altri organi. Quindi questa è una prima divisione importante per quanto riguarda la sede di origine tra i linfomi di Hodgkin e i linfomi non- Hodgkin. Inoltre spesso nei linfomi non-Hodgkin c'è un interessamento midollare e l'interessamento midollare se noi consideriamo la stadiazione che abbiamo visto per il linfoma di Hodgkin, se un paziente ha un interessamento midollare automaticamente diventa IV Stadio. L'interessamento di un organo extralinfonodale lo fa diventare IV Stadio. Il IV Stadio del linfoma di Hodgkin non ha la stessa valenza del IV Stadio del linfoma nonHodgkin. Il IV Stadio del linfoma di Hodgkin indica proprio lo stadio finale, lo stadio più avanzato della malattia linfomatosa, mentre una localizzazione midollare e quindi un IV Stadio di un linfoma non- Hodgkin potrebbe non indicare una malattia così grossa e così avanzata perché la malattia potrebbe fin dall'inizio interessare il midollo, ma poi determinare una piccola quota di malattia proliferativa linfonodale. È un paziente che su un piano nosografico noi lo integriamo come IV Stadio per l'interessamento midollare, ma è un IV Stadio che non è così pesate sul quadro prognostico come un IV Stadio di un linfoma di Hodgkin. Proprio perché il linfoma non-Hodgkin tende a localizzarsi al di fuori dei siti linfonodali la definizione di stadio nel linfoma non-Hodgkin non ha la stessa valenza come la definizione di stadio del linfoma di Hodgkin perché il linfoma di Hodgkin abbiamo detto che si propaga per contiguità, da un linfonodo passa ad un altro linfonodo e così via e poi alla fine diventa disseminato anche ad altri organi, mentre il linfoma non- Hodgkin si propaga per via ematogena e c'è la possibilità di localizzazioni extralinfonodali senza che questo costituisca espressione di una malattia molto avanzata. Noi però per convenzione utilizziamo le stesse stadiazioni come quelle del linfoma di Hodgkin però ripeto un IV Stadio di un linfoma non-Hodgkin non ha la stessa valenza di un IV Stadio di un linfoma di Hodgkin. (slide) Questo che non è una condizione possibile nel linfoma di Hodgkin, stavolta è invece possibile nel linfoma non- Hodgkin, cioè è possibile che il paziente abbia localizzazioni linfonodali distanti tra di loro proprio perché la diffusione della malattia è una diffusione per via ematogena. Ciò non è previsto nel linfoma di Hodgkin dove appunto la propagazione della malattia è per contiguità. (slide) Questa è la sezione anatomo-patologica di un linfonodo che vi fa vedere come a parte la capsula, a parte il seno marginale del linfonodo, qui c'è in questa zona più chiara il cosiddetto centro germinativo che è circondato dal cosiddetto mantello ed infine dalla zona marginale, questa qui all'esterno. Questo è importante perché i linfomi possono originare da una di queste tre zone e a seconda di dove originano questi linfomi hanno un comportamento clinico anche differente. Vi ricordo che gli organi linfoidi primari sono il timo e il midollo: il timo per quanto riguarda i linfociti T, il midollo per quanto riguarda i linfociti B. Questi linfociti passano poi negli organi linfoidi secondari che sono rappresentati dalle tonsille, dal tessuto linfoide associato alle varie mucose, ai linfonodi, dal midollo stesso, dalla milza, dai linfonodi mesenterici, dalle placche di Peyer ecc.. Quindi cosa succede sul piano filogenetico di queste cellule? Queste cellule, parliamo di linfociti B, i quali nascono nel midollo, da qui passano nel sangue periferico e da qui poi nel linfonodo, nel linfonodo incontrano l'antigene, le cellule germinative che sono le cellule presentanti l'antigene e questi linfociti che hanno incontrato l'antigene vanno incontro ad un riarrangiamento VDJ che a seconda che sia un riarrangiamento utile per l'organismo oppure un riarrangiamento non utile, le cellule se non è utile vanno incontro a morte per apoptosi altrimenti diventano cellule memoria o diventano plasmacellule. La stessa progressione maturativa che hanno i B-linfociti ce l'hanno pure i T-linfociti per cui i Blinfociti sono “precursor”, “pre-B”, “B” e “plasmacellule” e lo stesso per quanto riguarda i linfociti.... ( si sente solo rumore per qualche secondo). Se noi vogliamo immaginare questi linfomi da dove originano, il punto anatomico da cui originano dobbiamo considerare che ci sono cellule che hanno lasciato il midollo e che sono cellule “vergini” perché non hanno incontrato l'antigene che vanno a finire nel centro germinativo del linfonodo dove incontrano le cellule presentanti l'antigene e quindi incontrano l'antigene e qui subiscono il processing dei geni delle immunoglobuline e ci possono essere anche mutazioni del gene bcl6 che è quello che regola in traffico di questi linfociti all'interno del centro germinativo, poi queste cellule diventano cellule di memoria o diventano plasmacellule. In generale i linfomi cosiddetti mantellari che sono i linfomi che stanno appunto nel “mantello” che sta a coprire il centro germinativo. Questi sono linfomi che originano da cellule che ancora non hanno incontrato l'antigene. Viceversa la stragrande maggioranza degli altri linfomi come il “linfoma follicolare”, il “linfoma di Burkitt”, il “linfoma diffuso a grandi cellule” originano dal centro germinativo. Infine ci sono linfomi che originano invece da cellule più mature che sono le cellule di memoria o le plasmacellule che sono fondamentalmente i “linfomi della zona marginale” cosiddetti, i “linfomi del tessuto linfoide associato alle mucose” e alcuni linfomi a grandi cellule. Se voi prendete i vecchi libri c'erano fondamentalmente quattro tipi di linfomi non-Hodgkin. La classificazione attuale dei linfomi non- Hodgkin non ci sta in due diapositive. Ci sono adesso delle classificazioni dei linfomi non-Hodgkin particolarmente estese, però voi non dovete sapere ogni entità nosografica. Quello che dovete conoscere è che esiste una chiave di lettura di queste classificazioni ed esiste un ordine mentale con il quale sono state definite e stilate queste classificazioni. Tutte queste classificazioni si basano su alcuni principi di base. I principi di base sono: − prima di tutto che esistono i linfomi che originano dai linfociti B e i linfomi che originano dal linfociti T. La classificazione basata sul tipo di linfociti, se originano dai linfociti B o dai linfociti T. − Il secondo criterio classificativo è quello basata sull'aspetto al microscopio di questo linfoma, se è un aspetto nodulare o follicolare o un aspetto diffuso, cioè se l'aspetto ricalca quello del linfonodo normale che è formato da tanti follicoli oppure l'architettura del linfonodo è del tutto scompaginata da questa diffusa proliferazione linfoide. Questo secondo criterio è molto importante, cioè è un linfoma che cresce in maniera follicolare, cioè ricalcando in qualche modo l'architettura del normale linfonodo o c'è uno scompaginamento diffuso dell'architettura linfonodale? Questo criterio è importante perché permette di differenziare i linfomi in linfomi cosiddetti nodulari o anche cosiddetti follicolari rispetto ai linfonodi cosiddetti diffusi. − L'altro criterio importante è la grandezza di queste cellule neoplastiche, se si tratta di piccoli linfociti o si tratta di grandi linfociti. Quindi piccoli linfociti con cromatina matura oppure grandi linfociti con cromatina lassa. Questi due aspetti sono importanti per definire l'aggressività della malattia, per cui usualmente, diciamo che è una regola abbastanza larga, abbastanza generale, ma prendiamola come regola generale. I linfomi che hanno un tipo di architettura cosiddetta follicolare o cosiddetta nodulare, cioè che hanno un crescita di tipo sovrapponibile a quella di un normale linfonodo sono linfomi che si comportano in maniera indolente mentre i linfomi che hanno una diffusa alterazione del linfonodo sono linfomi che si comportano in maniera aggressiva. Quindi questa prima distinzione. Follicolare vs Diffuso. Follicolare è indolente mentre diffuso è aggressivo. Seconda distinzione: piccole cellule vs grandi cellule. Piccole cellule configurano un linfoma indolente, grandi cellule configurano un linfoma aggressivo. Quindi fondamentalmente tutti i criteri che informano queste classificazioni sono basati su questi tre punti: − primo punto: se si tratta di linfociti B o T; − secondo punto: se si tratta di una proliferazione di tipo nodulare o diffusa; − terzo punto: se si tratta di una proliferazione di piccoli linfociti o di grandi linfociti Domanda di una collega: “Ma l'aggressività riguardo le cellule B o le cellule T?” Risposta: “ I linfomi T sono più aggressivi”. Tutte queste classificazioni vedete come nel tempo sono andate migliorando in termini di chiarezza e che abbiamo degli strumenti differenti. Mentre prima nel '60 avevamo soltanto la morfologia e quindi la classificazione prevedeva quattro tipi di linfomi ed era basata prevalentemente sulla morfologia, successivamente si è basata sia sulla morfologia che sul fenotipo, quindi la distinzione tra B e T linfociti e altre caratteristiche fenotipiche. Poi c'è stata la “Working Formulation” che ha aggiunto a queste due caratteristiche l'aspetto più clinico. Infine c'è la classificazione REAL/WHO che adesso ha subito dei riarrangiamenti, ma che fondamentalmente è presente dagli anni '90, almeno il primo abbozzo è stato fatto negli anni novanta. La classificazione è basata non solo sulla morfologia, non solo sul fenotipo, non solo sulla clinica ma anche sul genotipo, cioè mano a mano che abbiamo cominciato a conoscere quelle che sono le alterazioni geniche di queste patologie, abbiamo anche iniziato a classificarle sulla base di questi aspetti. Quindi la classificazione attuale prevede fondamentalmente una prima distinzione in: − neoplasie a cellule B; − neoplasie a cellule T. Poi prevede una distinzione in: − neoplasie che derivano dai precursori dei linfociti. I linfociti nella loro ontogenesi hanno una sede dove sono precursori, quindi timo e midollo e una sede dei cosiddetti organi linfoidi secondari, tutti gli altri. Quindi se queste neoplasie originano nel timo o nel midollo sono neoplasie che originano dai precursori del B-linfociti o dei T-linfociti. Se le neoplasie vanno ad originare dal tessuto linfoide cosiddetto periferico allora sono neoplasie B o T del tessuto linfoide periferico. Poi vi è la classificazione delle neoplasie soltanto dei linfociti B e delle neoplasie soltanto dei linfociti T. Ci sono anche ulteriori classificazioni. Chiaramente voi non dovete saperle ma dovete sapere che esistono delle classificazioni estremamente dettagliate ma che si rifanno a questi principi. La cosa importante vi dicevo prima è che ci sono alcune alterazioni genetiche che identificano alcuni linfomi ed oltre ad identificare alcuni linfomi almeno in parte ne spiegano la patogenesi. Questo è il caso ad esempio del linfoma di Burkitt che si associa sempre ad una traslocazione 8;14. Oppure il caso del linfoma follicolare che si associa quasi sempre ad una traslocazione 14;18. Il linfoma mantellare si associa ad una traslocazione 11;14. Il linfoma anaplastico a grandi cellule si associa ad una traslocazione 2;5. Cioè ci sono delle traslocazioni cromosomiche che fanno in modo che un “pezzo” di cromosoma va a finire in un altro cromosoma e viceversa. È proprio una traslocazione reciproca. Queste traslocazioni fondamentalmente possono essere di 2 tipi in linea di massima. Abbiamo noi delle traslocazioni in cui il “pezzo” di un gene va a legarsi al “pezzo” di un altro gene per creare un “gene di fusione” e di conseguenza una “proteina di fusione”. Questo lo vedremo, è tipico delle leucemie dove si crea una proteina di fusione, cioè una proteina anomala che non esiste in natura e che quindi avrà delle funzioni anomale che poi determineranno lo sviluppo di alcune patologie, in particolare le leucemie. Nel caso invece dei linfomi la condizione che più frequentemente si viene a creare è che la regione regolatoria di un pezzo di gene va a finire di regolare un altro pezzo di gene, cioè in questo caso non si crea una proteina di fusione ma si crea una condizione nella quale un gene regolatore in realtà non andrà a regolare più il suo gene normale, ma andrà a regolare un altro gene. Questo è quello che succede nelle traslocazioni dei linfomi. In particolare per esempio, se prendiamo come esempio il linfoma mantellare che si associa ad una traslocazione 11;14, se fate caso il 14 è quasi sempre implicato in queste traslocazioni. Quindi nella stragrande maggioranza delle traslocazioni che identificano i linfomi nonHodgkin c'è un coinvolgimento del 14. Ma perché questo 14 è sempre implicato? Perché nel 14 ci stanno i geni nelle immunoglobuline. Se stanno lì i geni delle immunoglobuline, il linfocita B (perché parliamo di linfociti B esclusivamente) di “mestiere” fa quello che deve produrre le immunoglobuline, quindi capite bene che questo gene è particolarmente importante nel linfocita B e il promotore del gene delle immunoglobuline è un promotore molto attivo perché nei linfociti B il gene che codifica per immunoglobuline è sempre acceso, quindi c'è un promoter che gli “dice” sempre di stare acceso. Questo promoter che regola in questo caso il gene delle immunoglobuline va a finire a regolare un altro gene e quindi dà l'ordine di stare sempre accesso ad un altro gene che non è più quello delle immunoglobuline. Nel caso specifico della traslocazione 11;14 il gene che viene stimolato dal promoter del gene delle immunoglobuline è il gene bcl-1 che è un gene che regola il ciclo cellulare e quindi queste cellule hanno un'alterazione del ciclo cellulare che è almeno in parte responsabile della loro responsabilità proliferativa. Lo stesso succede nella traslocazione 14;18 che è invece patognomonica del linfoma follicolare. In questo caso il gene che viene sempre stimolato è il bcl2 che è un gene anti-apoptotico per cui la cellula avendo sempre questo bcl-2 accesso non muore, per cui questo è un meccanismo di accumulo cellulare, non tanto di proliferazione ma di accumulo, tanto è che il linfoma follicolare è un linfoma indolente, non è un linfoma aggressivo, cioè le cellule del linfoma follicolare non è che crescono più rapidamente, piuttosto le cellule non muoiono, la loro normale emivita viene alterata dal fatto che gli blocchiamo i meccanismi della apoptosi e quindi sono patologie che crescono lentamente proprio per l'accumulo, per la mancanza dei processi apoptotici. Al contrario invece per esempio nel linfoma di Burkitt c'è la traslocazione 8;14 e in questo caso il gene che viene coinvolto è il c-myc. Il c-myc è un gene che serve per la proliferazione cellulare. Il c-myc normalmente quando la cellula deve proliferare si accende e la cellula prolifera, quando la cellula smette di proliferare si spegne il c-myc. In questo caso il c-myc sarà sempre acceso e la cellula prolifera in maniera indipendente da tutti i meccanismi di controllo della crescita cellulare. In questo caso il linfoma di Burkitt non sarà un linfoma a lento decorso come il linfoma follicolare, ma sarà un decorso rapidamente progressivo, rapidamente evolutivo, proprio perché sono accesi quei meccanismi che servono alla proliferazione cellulare. Queste alterazioni che noi vediamo sono quelle grossolane. In tutti gli altri linfomi semplicemente non sono descritte queste traslocazioni cromosomiche perché sono così sottili che la citognetica convenzionale non riesce ad evidenziarle. Ma sempre di più escono report della letteratura che dicono che se noi andiamo a fare delle analisi più sofisticare del DNA vediamo sempre di più delle traslocazioni che sono criptiche alla citogenetica convenzionale ma che sono invece evidenziate da metodiche di biologia molecolare o di imaging molecolare più profondo che ci permettono di evidenziare che praticamente tutti i pazienti con processi linfoproliferativi hanno una qualche alterazione del DNA che giustificano il motivo della loro proliferazione. Quali sono le vie che portano alla trasformazione di una cellula normale in una cellula maligna, di un B-linfocita normale ad un B-linfocita linfomatosa? − soppressione dell'apoptosi; − alterazione della regolazione del ciclo cellulare; − l'alterazione del controllo della differenziazione; − alterazioni della capacità di queste cellule di homing, termine inglese con cui si indica la capacità di queste cellule di andare a localizzarsi in determinati organi e determinati tessuti. Ecco perché i linfomi non-Hodgkin non sono soltanto linfomi che interessano i linfonodi ma spesso interessano altri tessuti oltre al tessuto linfonodale perché c'è una alterazione della capacità di homing di queste cellule, non è che vanno a localizzarsi soltanto nei linfonodi dove le cellule normali vanno a localizzarsi perché queste cellule hanno una homing linfonodale specifica. Queste cellule perdono la capacità di homing per cui vanno a disseminarsi anche in altri tessuti che non sono il tessuto linfonodale stesso. (slide) Qui è localizzata una possibilità di una linfomagenesi e progressione. Esistono i linfomi che hanno una bassa capacità di crescita dove appunto il meccanismo più importante nella genesi del linfoma è l'inibizione dell'apoptosi e il prototipo di questo linfoma è il linfoma follicolare dove appunto a causa della traslocazione 14;18 c'è un aumento del bcl2 che è il gene anti-apoptotico e quindi c'è l'inibizione dell'apoptosi. Lo stesso vale anche per i linfomi MALT, che sono i linfomi che originano dal cosiddetto tessuto MALT (“Mucose-Associated Lymphoid Tissue”, cioè Tessuto Linfoide Associato alla Mucosa). Per esempio questi linfomi originano nello stomaco perché nello stomaco c'è questo tessuto linfoide associato alla mucosa che può degenerare in senso linfomatoso. In questo caso vi sono traslocazione 1;14 e la traslocazione 15;18. Entrambe le condizioni provocano o un'alterazione del gene bcl-10 (?) o la formazione di un gene ibrido. Questa è una delle rare eccezioni in cui si forma un gene libero che porta ad una iperattivazione di NFKB che è una sorta di “motore” cellulare che serve alla proliferazione, che serve all'angiogenesi, che serve alla produzione di alcune citochine ecc.. e quindi tutto questo può degenerare verso un linfoma più aggressivo, condizione questa che succede in clinica. Quello che noi vediamo più spesso sono linfomi indolenti che in alcuni pazienti accumulano altre alterazioni genetiche e da linfoma indolente diventano linfoma aggressivo. Tutta questa condizione è dovuta o comunque peggiora una instabilità genomica così queste cellule acquisiscono altre alterazioni genomiche che sono poi responsabili della progressione. Viceversa i linfomi che nascono già come linfomi aggressivi sono quei linfomi dove c'è una attivazione della proliferazione. Il prototipo è il linfoma di Burkitt in cui c'è la traslocazione 8;14 che attiva il c-myc, ma anche i linfomi cosiddetti diffusi a grandi cellule dove c'è l'interessamento di alcuni cromosomi ma soprattutto c'è una mutazione del gene bcl-6. Il gene bcl6 è quello che regola il centro germinativo, quindi queste mutazioni a carico del bcl6 fanno sì che aumenta il bcl6 o comunque è un bcl6 anomalo che favorisce la proliferazione incontrollata di queste cellule all'interno del centro germinativo. Poi mano a mano che queste cellule acquisiscono delle altre alterazioni molecolari, ad esempio deficit di NPM2(?), la delezione del p53. Sapete che il p53 è il guardiano della cellula, manda a morte la cellula qualora ci sia un danno genomico per cui appena c'è un danno genomico si attiva la via del p53 e si ha l'apoptosi della cellula che permette alla cellula di morire e non trasmettere alla cellula quel danno genomico. Se manca la p53 questi danni genomici si accumulano e quindi determinano un fenotipo progressivamente più maligno della cellula. Detti questi rudimenti di biologia molecolare che riguardano i linfomi passiamo alla clinica dei linfomi. Il prof mostra un grafico a torta sulla casistica dei linfomi: i linfomi a cellule T rappresentano un 10%, tutto il resto è rappresentato da linfomi a cellule B e di questi linfomi a cellule B una grossa fetta è rappresentata da quello che è il prototipo dei linfomi indolenti che è il linfoma follicolare e da quello che è il prototipo dei linfomi aggressivi che è il linfoma diffuso a grandi cellule. Questi sono i due più frequenti tipi di linfoma: il linfoma follicolare e il linfoma diffuso a grandi cellule. Poi esiste tutta una serie di altri linfomi che vedremo successivamente. Ma fondamentalmente, senza andare nel dettaglio delle singole entità nosografiche quello che a voi importa sapere è che esistono linfomi indolenti e linfomi aggressivi. I linfomi indolenti sono i linfomi cosiddetti a basso grado di malignità, i linfomi aggressivi sono quelli ad alto grado di malignità. Vi ripeto che il prototipo del linfoma indolente è il linfoma follicolare. Il prototipo del linfoma aggressivo è il cosiddetto “linfoma aggressivo a grandi cellule”. Le caratteristiche anche di storia naturale di queste malattie sono differenti perché i linfomi indolenti crescono ma molto lentamente mentre i linfomi aggressivi crescono rapidamente ma hanno anche una diversa risposta al trattamento e una diversa storia naturale. I linfomi indolenti crescono lentamente, rispondono al trattamento ma ricadono e alla fine non sono guaribili (not curable). Quindi i linfomi indolenti seppure abbiano una storia molto più lenta e più lunga alla fine risultano inguaribili. Viceversa i linfomi aggressivi che hanno una clinica molto più rapida, una storia naturale molto più aggressiva alla fine sono quelli più sensibili alla chemioterapia, rispondono molto bene alla chemioterapia, non ricadono e quindi alla fine guariscono. Quindi abbiamo paradossalmente da un lato un linfoma indolente, molto tollerato dal paziente, che dà meno sintomi ma che non guarisce, dall'altro un linfoma molto più aggressivo che però guarisce se trattato. Quindi quali sono i linfomi indolenti? Il prototipo è il linfoma follicolare ma esiste tutta una serie di altri linfomi indolenti. Il linfoma follicolare è quello più importante, quello più comune, colpisce soprattutto pazienti di età superiore a 40 anni, è caratterizzato da queste piccole cellule. Vi ricordo i criteri che formano la classificazione istologica dei linfomi. Oltre ad un criterio se sono B linfociti o T linfociti, il secondo criterio è il tipo di alterazione architetturale del linfonodo, nodulare (o follicolare) o diffuso. I linfomi follicolari sono indolenti, i linfomi diffusi sono aggressivi. Questo calza con quello che stiamo dicendo ora, cioè il linfoma follicolare che ha quel tipo di proliferazione di tipo follicolare è un linfoma è un linfoma indolente, viceversa il linfoma diffuso a grandi cellule che altera completamente l'architettura del linfonodo è il linfoma aggressivo. Il linfoma follicolare prevalentemente è formato da piccole cellule. Queste piccole cellule configurano un andamento indolente, viceversa il linfoma diffuso a grandi cellule per definizione è appunto formato da grandi cellule. Nel caso specifico il linfoma follicolare è caratterizzato da questa traslocazione 14;18 che fonde il gene delle immunoglobuline con il gene bcl-2. La conseguenza è che c'è una maggiore espressione della proteina bcl-2 la quale inibisce l'apoptosi. C'è questo meccanismo patogenetico peculiare del linfoma follicolare di traslocazione 14;18 che determina il passaggio del promoter del gene delle immunoglobuline vicino al gene bcl-2, quindi iperproduzione di proteina bcl-2, blocco dell'apoptosi. (slide) Architettura di un linfoma follicolare: disordinata proliferazione di questi follicoli che però pur essendo disordinati, pur essendo un po' più grandi del normale però mantengono questa architettura follicolare. Ancora una volta vi ricordo che questo dipende da una traslocazione 14;18 per cui un pezzo di cromosoma 14 va a finire sul 18 e viceversa un pezzo del 18 va a finire sul 14. Tutto questo provoca una iperproduzione della proteina bcl-2 che usualmente è localizzata nei mitocondri, nel reticolo endoplasmatico ecc e la cui funzione fisiologica è quella di conferire longevità alle cellule e quindi c'è un prolungamento della sopravvivenza per blocco dell'apoptosi. Il bcl-2 è una proteina che tutti quanti noi abbiamo, non è che bcl-2 è la proteina che determina la trasformazione del linfoma da cellula normale a cellula linfomatosa. La proteina bcl-2 ce l'hanno tutte le cellule che vivono a lungo. Non c'è usualmente nel centro germinativo, ma c'è nella zona del mantello dove ci stanno le cellule memoria. Se noi prendiamo un linfonodo normale e facciamo una immunoistochimica per bcl-2 noi vedremo che bcl-2 è presente sulle cellule del mantello perché sono cellule che durano a lungo, che hanno una longevità. Viceversa nel centro germinativo, che è un centro in cui c'è una rapida proliferazione e in cui le cellule vanno incontro all'apoptosi perché appunto tutte quelle cellule che hanno un tipo di riarrangiamento non funzionale vanno incontro ad apoptosi, quindi qui c'è poco bcl-2 in condizioni normali. Invece nel linfoma follicolare succede il contrario e questo è un parametro molto importante per l'anatomo-patologo per dire se il paziente ha un linfoma oppure no perché il linfoma follicolare in qualche modo, anche se in maniera disordinata riproduce la stessa architettura del linfoma normale quindi spesso negli anni in cui non c'era l'immunoistochimica era difficile fare una analisi differenziale tra le linfoadeniti reattive dove c'è una iperplasia follicolare reattiva a qualche fatto infiammatorio rispetto al linfoma follicolare, perché in ambedue i casi c'è un aumento di questi follicoli. Non è che la cellula di un linfoma follicolare sia molto differente da un linfocita normale, si tratta sempre di linfociti piccoli, con una cromatina compatta, allora uno dei parametri più importanti che vengono in ausilio all'anatomo-patologo per definire una diagnosi di linfoma follicolare è quello della bcl-2. Facendo l'immunoistochimica per bcl2 si vede come che nella linfoadenite reattiva i centri germinativi sono bcl-2-negativi mentre la zona di contorno, la zona mantellare è bcl-2-positiva perché lì ci stanno le cellule della memoria. Se il centro germinativo è pieno di cellule bcl-2-positive che si colorano in marrone, quelle sono cellule linfomatose. Quindi questo è il quadro immunoistochimico differenziale tra una linfoadenite reattiva e il linfoma follicolare. Questo è comunque un discorso molto generale perché le cose non stanno sempre così e potete vedere come ad esempio alcuni linfomi follicolari sono negativi per bcl-2 all'interno del centro germinativo, ci sono delle eccezioni ovviamente però vedete come nel caso di una iperplasia follicolare benigna è negativo in tutti i pazienti, positivo in nessun paziente. La negatività per bcl-2 in un linfonodo con iperplasia follicolare e la positività nel follicolo di un linfonodo con linfoma follicolare. Quindi il bcl-2 è uno dei marcatori più importanti che l'anatomo-patologo utilizza adesso per fare la diagnostica di linfonodo follicolare. Però il bcl-2 non lo utilizziamo soltanto per la diagnostica, lo utilizziamo anche per la valutazione della malattia Malattia Minima Residua, cioè il vantaggio di aver identificato questa alterazione citogenetica e poi molecolare di bcl-2 ci permette di avere un'arma in più non soltanto per la diagnosi, ma ci permette di avere un'arma in più anche nella valutazione del residuo di malattia che è rimasta dopo la terapia perché la morfologia avete visto non è così differente tra una condizione benigna e una condizione maligna. Se noi andiamo per esempio a fare un prelievo di midollo a questi pazienti (spesso il midollo è contaminato da malattia linfomatosa), quindi noi preleviamo il midollo, sulle cellule del midollo facciamo bcl-2, facciamo la valutazione del riarrangiamento genico che ha portato all'iperespressione di bcl2 e lo troviamo positivo. Facciamo la terapia poi e ricontrolliamo questo riarrangiamento genico. Questo ci permette di avere un potere di risoluzione di gran lunga superiore rispetto alle metodiche di morfologia, perché se noi questo midollo lo vediamo solamente con la morfologia il potere di risoluzione è grossomodo del 5%, cioè noi nella migliore delle ipotesi riusciamo a dire che c'è una localizzazione di malattia linfomatosa quando c'è almeno il 5% di linfociti che a noi ci sembrano linfomatosi (5 su 105). invece le metodiche di biologia molecolare hanno un potere di risoluzione di 1 cellula malata su 100 000 cellule sane, quindi siamo passati da una capacità di risoluzione del 5% ad una capacità di risoluzione che è di 1:100 000 (105). È differente la capacità di vedere la malattia residua che abbiamo attualmente rispetto a quella che avevamo dieci anni fa. Questo ci permette anche di poter modulare la terapia in maniera differente e, non dico che ci stiamo riuscendo, ma in molti casi quel dogma che il linfoma follicolare necessariamente deve ricadere adesso non è più così, perché prima ricadeva perché noi non riuscivamo ad andare al di sotto di una “soglia di dimostrabilità” di un linfoma residuo. Quando noi facevamo un trattamento ad un paziente con linfoma follicolare gli facevamo la TAC, la biopsia del midollo e non vedevamo più niente, è vero che a livello macroscopico non vedevamo più niente, ma sicuramente è perché la nostra soglia di “detection” della malattia era molto alta. Adesso che abbiamo abbassato questa soglia vediamo in questi pazienti che c'è un residuo di malattia e su quel residuo di malattia possiamo intervenire e questa è probabilmente la ragione per cui adesso molti pazienti non ricadono, perché otteniamo delle risposte molecolari, cioè delle risposte che hanno una qualità di gran lunga superiore rispetto a quelle che noi ottenevamo prima quando avevamo soltanto degli strumenti grossolani di valutazione della malattia. Quindi la cosa importante è che bcl-2 ci permette non soltanto di migliorare le nostre armi diagnostiche, ma ci permette anche di migliorare le nostre armi di valutazione della malattia residua. Allora quali sono le caratteristiche cliniche di questi linfomi a basso grado? I linfomi a basso grado sono linfomi vi dicevo che determinano un ingrossamento lento e quasi sempre asintomatico dei linfonodi, linfonodi che hanno una consistenza gommosa, mobili, raramente sono linfonodi fissi sui piani circostanti. Vi dicevo la lezione scorsa che questi linfonodi possono avere un decorso a “fisarmonica” cioè possono ingrossarsi e ridursi spontaneamente oppure anche a seguito di processi infettivi che liberano citochine che determinano una riduzione di questi linfonodi. Interessano i linfonodi, la milza, il fegato, il midollo osseo, spesso ci sono anche interessamenti extranodali, tipo la pleura, il polmone, la pelle, la mammella, il tratto gastroenterico e usualmente si presentano in stadio avanzato. Quello che vi dicevo prima, se noi adoperiamo la definizione di stadio, la stessa classificazione del linfoma di Hodgkin, una classificazione extralinfonodale configura già uno stadio avanzato. Però noi vediamo nei linfomi follicolari pazienti molto spesso che sono etichettati come stadio IV, stadio avanzato, ma in realtà hanno poca malattia, hanno magari pochi linfonodi ingrossati e un interessamento midollare che li etichetta come stadio IV ma certamente non sono pazienti che hanno una...... (51:30), per cui la stragrande maggioranza di questi pazienti si presenta in stadio avanzato. Hanno una lunga sopravvivenza mediana caratterizzata da ricadute multiple, cioè rispondono al trattamento e poi ricadono, rispondono e poi cadono di nuovo. Anche se questo negli ultimi anni lo stiamo un po' aggiustando. Poi hanno la particolare predisposizioni alle malattie autoimmuni. Questo non è che l'abbiamo capito bene noi perché la prima cosa a cui si pensa è che essendo questi dei linfomi cioè delle patologie neoplastiche che interessano i linfociti che producono immunoglobuline, essendoci questa espansione clonale dei linfociti B che sono neoplastici producono tante immunoglobuline e poi queste immunoglobuline producono un fattore autoimmune. Questa sarebbe la spiegazione logica. Il problema è che queste immunoglobuline che poi sono responsabili della autoaggressività, dell'autoimmunità non sono le stesse immunoglobuline prodotte dal clone neoplastico, ma sono immunoglobuline prodotti da linfociti normali, da linfociti che non sono coinvolti dal processo neoplastico. L'ipotesi è che queste patologie determinano una alterazione dei cosiddetti linfociti T regolatori che di “mestiere” servono proprio a regolare, ad evitare l'espansione di cloni autoimmunitari e quindi abbassandosi in qualche modo la sorveglianza di questi cloni autoimmunitari perché si abbassano i linfociti T regolatori c'è una maggiore probabilità che vengano fuori dei cloni autoimmuni che non sono cloni neoplastici, sono linfociti assolutamente normali ma che producono poi immunoglobuline che poi vanno ad essere responsabili dei fenomeni autoimmuni e il bersaglio preferiti di questi cloni autoimmunitari sono i globuli rossi per cui questi pazienti hanno spesso una anemia emolitica autoimmune. Quando noi facciamo diagnosi di anemia emolitica autoimmune, quindi sviluppo di anticorpi che possono essere anticorpi caldi, anticorpi freddi ecc, il dovere che abbiamo è andare a cercare la malattia linfoproliferativa perché gran parte delle anemie emolitiche autoimmuni nascondono o sono un epifenomeno di una malattia linfoproliferativa, più spesso di una malattia linfoproliferativa di tipo indolente, a basso grado di malignità. (slide) Questa è una curva di sopravvivenza degli anni scorsi e vi fa vedere come inesorabilmente questi pazienti hanno una sopravvivenza che decade, questa curva non ha un plateaux, ha un andamento inesorabilmente verso la morte. La mediana di sopravvivenza di questi pazienti è di otto anni e sono pazienti che sopravvivono anche 25 anni e più, quindi per dire che non sono patologie aggressive ma sono patologie che non guariscono. Casistiche di posti considerati i migliori al mondo mostrano gli stessi risultati. C'è stato negli anni un lieve miglioramento, ma sempre relativo, l'andamento della curva è sempre lo stesso. Un'altra possibilità, come dicevo prima, è che i linfomi follicolari diventino diffusi e da un linfoma a basso grado di malignità si passi ad un linfoma ad alto grado di malignità e questo dipende dal fatto che c'è un accumulo di alterazioni citogenetiche e molecolari che permettono ad un linfoma a basso grado di malignità di diventare un linfoma ad alto grado di malignità proprio perché accumula alterazioni molecolari e a quel punto non è solo la bcl2 che comanda ma sono tante altre alterazioni che vedremo. Domanda di una collega: “Quindi l'aggressività aumenta nel momento in cui iniziano a diventare diffusi?” Risposta: “Esatto”. I pazienti non vanno tutti allo stesso modo. C'è una scala gerarchica prognostica che identifica pazienti che hanno una malattia più blanda rispetto a pazienti che hanno una malattia più aggressiva e non può essere la sola stadiazione come succede per il linfoma di Hodgkin dove la stadiazione ha un suo significato prognostico importante. Qui la stadiazione ha un significato ma non è così importante. Per cui di recente sono state sviluppate delle altre classificazioni che permettono di prevedere la prognosi di questi pazienti e queste classificazioni nel caso dei linfomi follicolari sono basati sulla età, età superiore o inferiore a 60 anni, i valori di emoglobina, i valori di LDH, lo stadio, stabilito come vi ho fatto vedere secondo la ….... e il numero di localizzazione nodali, cioè di linfonodi coinvolti. Tutto questo permette di differenziare i pazienti in gruppi di rischio: − Il gruppo di rischio buono è quello che ha uno o nessun fattore di rischio e la sopravvivenza a 10 anni è del 71%. − Quelli che hanno un rischio intermedio con 2 fattori. − Quelli che hanno rischio scarso (?) (probabilmente intendeva “prognosi” scarsa) con più di tre fattori. Vedete che la sopravvivenza a 10 anni è differente nei tre gruppi, 71%, 51% e ..%. Sono tutti linfomi follicolari trattati nello stesso modo con però una sopravvivenza differente a seconda che il paziente si trovi nel gruppo a prognosi buona, intermedia o scarsa. In questi pazienti noi possiamo adottare anche un atteggiamento di tipo attendistico proprio perché sono pazienti che non guariscono, proprio perché sono pazienti che rispondono comunque a successive terapie esiste una scuola di pensiero soprattutto americana che dice che visto che questi pazienti non li facciamo guarite e visto che questa malattia è talmente indolente che spesso la diagnosi la facciamo per caso perché il paziente si presenta da noi perché si è accordo di un “linfonodino” latero-cervicale ma non ha nessun sintomo come pazienti che stanno bene, in questi casi è possibile adottare un atteggiamento di tipo “watch and wait” (guarda e aspetta), cioè trattiamo questa malattia solo quando dà una compromissione dell'organismo. Questo atteggiamento ha permesso di vedere che alcuni di questi pazienti vanno incontro a remissione spontanea. Sono stati fatti degli studi in cui i pazienti sono stati solamente osservati e si è visto che una quota di pazienti che non è indifferente, intorno al 20%, hanno una riduzione dei linfonodi, addirittura in qualche paziente spariscono i linfonodi per poi ripresentarsi magari nel tempo. Oppure ci sono dei lunghi periodi di stabilità. Noi abbiamo diversi pazienti, non tanto con il linfoma follicolare perché il linfoma follicolare tende più spesso a peggiorare, ma altri linfomi indolenti come i cosiddetti linfomi della zona marginale che sono stabili per un lungo periodo di tempo, hanno un loro linfonodo che sta lì, fermo che non si muove, non danno nessuna compromissione all'organismo, non danno nessuna sintomatologia per cui per questi pazienti è accettato che non si faccia più una terapia. Questo è un concetto che è difficile da accettare dal paziente e ancora meno dal medico curante. Quando io dico al paziente: “Lei ha un linfoma” oppure “Lei ha una leucemia a basso grado e non deve essere curato” succede a volte che il medico curante invita il paziente a cambiare ematologo. Questo perché non si rende conto che alcune condizione non hanno necessità di non essere trattate. Se noi abbiamo un trattamento che fa guarire il paziente allora dobbiamo assolutamente trattare anche la malattia piccola, ma se il trattamento non fa guarire il paziente si danno farmaci senza utilità in termini di sopravvivenza perché è dimostrato che il “watch and wait” non preclude e non riduce l'aspettativa di sopravvivenza di questi pazienti. Il prof ammette che il suo atteggiamento personale è quasi sempre interventistico, non applica il “watch and wait” a tutti i pazienti con linfoma indolente perché ci sono in alcune condizioni pazienti giovani, pazienti che non riescono a convivere con l'idea di avere una malattia neoplastica. Ma soprattutto perché noi abbiamo delle armi terapeutiche di cui vi dirò successivamente che fanno sì che magari stiamo cambiando la storia naturale di queste malattie e non sono solo armi terapeutiche ma sono anche armi diagnostiche come vi dicevo prima, cioè la possibilità di identificare un residuo di malattia molto piccolo che ci permetti di andare oltre quello che si faceva prima. Cioè se adesso abbiamo pazienti che non hanno più malattia in termini di TAC, PET, biopsia midollare, ma che hanno la malattia soltanto molecolare, solo bcl-2 positiva che noi non vediamo se non attraverso questi esami di biologia molecolare, lì possiamo intervenire con ulteriore terapia, quindi abbassare ulteriormente il carico di malattia residua e prospettare per questi pazienti, se non una guarigione, ma per lo meno una sopravvivenza più lunga rispetto a quella prospettata prima quando non avevamo queste informazioni. Quindi abbiamo maggiori possibilità diagnostiche, maggiori possibilità di vedere una malattia residua, maggiori possibilità terapeutiche. Quindi sta cambiando un po' il nostro atteggiamento nei confronti di questi linfomi indolenti che prima venivano considerati semplicemente linfomi inguaribili, quindi inutile dare tossicità al paziente se il paziente e sta bene, non ha progressione della malattia. Adesso le cose cambiano un pochettino per cui questo atteggiamento è un pochettino ridotto, ma continua ad essere presente in alcuni pazienti. Uno dei farmaci nuovi che abbiamo a disposizione è rappresentato dagli anticorpi monoclonali, in particolare l'anticorpo monoclonale diretto contro il CD20 che è un antigene presente sulla superficie dei linfociti B e che è in grado di esercitare quella che noi chiamiamo la “immunoterapia”, quindi effetto tossico su questi linfociti B e questo ha potenziato significativamente l'efficacia della nostra terapia. Domanda di una collega: “Lei ha detto che la diagnosi spesso si fa per caso. Ma quando non si fa per caso, perché si fa? Cioè qual è il campanello d'allarme che deve dire al medico che questo paziente deve fare una biopsia? Perché la sintomatologia è comunque blanda.” Risposta: “La sintomatologia clinica nella stragrande maggioranza dei casi non c'è. Ci possono essere casi, ma sono rari, in cui c'è una sudorazione notturna, una febbricola, un dimagrimento che sono i tipici sintomi delle malattie linfoproliferative, però in questi casi di linfomi a basso grado usualmente non vi sono. Usualmente quello che noi vediamo sono pazienti con adenopatie multiple di una malattia che nasce usualmente già discriminata, sono pazienti che hanno linfonodi latero-cervicali, ascellari, inguinali, spesso c'è una splenomegalia e questo ci permette spesso di fare la diagnosi. A volte facciamo la diagnosi ancora più tardivamente, quando il paziente diventa anche anemico, diventa piastrinopenico, ma questi sono condizioni più... Ma spesso il motivo che porta il paziente alla nostra osservazione sono le linfoadenopatie multiple o isolate” Domanda della collega: “Ma questo succede in ambito ospedaliero?” Risposta: “Cioè il paziente si accorge, che so, facendosi la barba che trova dei linfonodi laterocervicali, oppure facendosi la doccia sente i linfonodi ascellari. Queste sono le condizioni che portano il paziente dal medico curante per quanto riguarda per la valutazione di questi linfonodi”. Lezione 8 L'altra volta abbiamo parlato dei linfomi non-Hodgkin e vi ricordo di tenere a mente quale è il punto dove nascono i linfomi non-Hodgkin che è il centro germinativo. Per cui ci sono i linfomi che originano dal centro germinativo che sono la maggior parte, da linfociti che stanno nel centro germinativo e che sono la maggior parte e linfomi che possono originare pre-centro germinativo o post-centro germinativo. Ricordiamo che esistono i linfomi a cellule B e linfomi a cellule T e che le classificazioni attuali prevedono non soltanto la morfologia, ma anche l'immuno-fenotipo, ma anche le informazioni cliniche e il genotipo. Se ricordate abbiamo distinto i linfomi in linfomi a cellule B e linfomi a cellule T e a seconda che interessano i precursori o le cellule più mature si distinguono neoplasie linfoidi dei precursori o delle cellule più mature. Vi ricordate che abbiamo parlato tanto delle traslocazioni, in particolare delle traslocazioni che interessano il cromosoma 14 perché lì ci sta il gene delle immunoglobuline e abbiamo parlato di tutte le principali alterazioni molecolari che sono responsabili della genesi dei linfomi, in particolare per esempio il bcl-2 che ci serve sia per la diagnosi che il follow-up dei pazienti con linfoma follicolare e che deriva da questa traslocazione (14;18) che iperesprime bcl-2. Parlando dei linfomi follicolari abbiamo visto come sono dei linfomi che almeno con le vecchie terapie sono non guaribili anche se hanno un andamento molto indolente. Linfomi aggressivi Passiamo adesso ai linfomi aggressivi. I linfomi aggressivi sono rappresentati soprattutto dal linfoma diffuso a grandi cellule. Poi c'è una serie di altri linfomi aggressivi, ma il linfoma diffuso a grandi cellule è sicuramente il prototipo dei linfomi aggressivi. (slide) Come vedete in questa torta di distribuzione della frequenza dei linfomi vedete che una bella fetta di linfomi è data da questi linfomi aggressivi indicati come DLCL (che sta per Diffuse Large Cell Lymphoma cioè Linfoma Diffuso a Grandi Cellule). Se ricordate quando abbiamo parlato delle classificazioni anatomo-patologiche una delle chiavi di lettura di queste classificazioni era il tipo di alterazione architetturale del linfonodo, se follicolare o diffusa e dicevamo che quelli follicolari sono indolenti a basso grado di malignità mentre quelli diffusi sono ad alto grado di malignità, quindi già l'aggettivo “diffuso” ci indica che è ad alto grado di malignità. L'altro parametro che abbiamo visto la volta scorsa era la grandezza delle cellule, se sono cellule piccole o sono cellule grandi. Visto che stiamo parlando di linfoma diffuso a grandi cellule anche questo è un elemento che ci dice appunto come si tratta di una patologia aggressiva, di un linfoma ad alta malignità. I linfomi aggressivi sono caratterizzati dal fatto che i linfonodi crescono molto rapidamente, sono linfonodi di consistenza gommosa, sono mobili ma duri alla palpazione e talvolta possono essere eritematosi e anche dolenti. Spesso sono presenti delle masse che noi chiamiamo “bulky mass”, cioè una grossa massa di malattia e identifichiamo come “bulky” quelle tumefazioni linfonodali, spesso sono agglomerati di linfonodi, che hanno un diametro superiore a 10cm e queste spesso le vediamo sia a livello mediastinico sia a livello mesenteriale. In genere i sintomi di questi linfomi sono legati proprio a questa rapida espansione dei linfonodi, contrariamente a quello che succede nei linfomi indolenti, in particolare nel linfoma follicolare o nel linfoma della zona marginale, nel caso dei linfomi aggressivi queste masse crescono così rapidamente che danno sicuramente molto fastidio al paziente. Oltre a questo ci sono i segni costituzionali del linfoma: perdita di peso, sudorazione notturna e la febbre. Intervengono in pratica a tutte le età, sono più comuni però dopo i 60 anni. Frequentemente interessano i siti extranodali quale per esempio il tratto gastroenterico, il fegato, la pelle ecc.. Più spesso sono in stadio non avanzato ma in stadio limitato, la localizzazione in stadio limitato è molto più frequente rispetto a quella che vediamo nei linfomi non-Hodgkin indolenti perché nei linfomi non-Hodgkin indolenti spesso questi pazienti nascono in stadio avanzato. Questi pazienti hanno una potenzialità di guarigione più elevata rispetto ai pazienti con linfoma indolente. Fatta una diagnosi di linfoma non-Hodgkin come vi dicevo l'altra volta dobbiamo fare la stadiazione, cioè dobbiamo sapere un po' la mappa di questa malattia e la mappa si fa facendo ovviamente la biopsia della lesione che ci permette di fare la diagnosi con un'attenta storia dei sintomi B, come abbiamo detto prima, ossia la febbre, la sudorazione notturna, il dimagrimento. Visitare il paziente e andare a guardare tutte le possibili zone di localizzazione della malattia e in particolare tutte le stazioni linfonodali del collo, delle ascelle, degli inguini, ma anche il fegato e la milza e non dimenticate di andare a guardare il Waldeyer che può essere sede di localizzazione di malattia. Fare una valutazione laboratoristica con gli esami del sangue che comprendono l'emocromo completo, LDH e la β2-microglobulina che sono due aspetti che indicano il grado di proliferazione, sono strettamente correlati alla rapidità della proliferazione del linfoma. Andare a vedere bene la funzione renale e la funzione del fegato e poi chiaramente fare degli studi di imaging che ci dicono qualcosa di più rispetto al semplice esame obiettivo. Usualmente facciamo una TAC. La scintigrafia con Gallio che si faceva una volta ora in pratica è stata sostituita dalla PET. Quello che si fa è soprattutto una valutazione del midollo con una biopsia osteo-midollare perché al momento non esistono immagini di imaging che ci permettono di identificare localizzazioni osteo-midollari di malattia che sono piuttosto frequenti. Vi ricordo la stadiazione dei linfomi: − Stadio I: una singola stazione linfonodale; − Stadio II: due o più stazioni dallo stesso lato del diaframma; − Stadio III: due o più stazioni da ambedue i lati del diaframma; − Stadio IV: localizzazioni extralinfonodali. Quando andiamo a visitare il paziente, oltre a visitarlo, dobbiamo cercare di definire il suo stato di salute, cioè come sta il paziente e chiaramente questa è una valutazione soggettiva e allora si sta cercando di definire in maniera più obiettiva possibile lo stato di validità del paziente. Ci sono diverse scale che vengono utilizzate per cercare di mettere in cartella un numero in modo tale che a quel numero corrisponda una situazione dello stato di validità del paziente. Diverse scale sono piuttosto complicate, quella che noi utilizziamo usualmente è una scala abbastanza semplice che divide i pazienti in 5 stadi ECOG PS (Performance Status) a seconda che il paziente : − non ha nessun sintomo, è pienamente attivo, abile a fare la sua normale vita e in questo caso lo definiamo come ECOG PS (Performal Status) Stadio zero (0). − Se invece ha una lieve riduzione della sua attività fisica, quindi per esempio non può salire le scale di corsa, però cammina ed è abile a svolgere il suo lavoro soprattutto se è un lavoro sedentario, un impiegato, allora in questo caso diamo una definizione di Stadio 1. − Quando è ancora capace di camminare, di badare a se stesso, ma non è capace di svolgere un lavoro anche sedentario, soprattutto se è in grado di camminare, di stare in piedi per più del 50% della sua giornata in questo caso gli diamo un punteggio 2. − Se invece è capace soltanto di una limitata capacità di autogovernarsi ed è costretto a letto per più del 50% della sua giornata diamo un punteggio 3. − Se invece è completamente disabile e confinato a letto per gran parte della giornata diamo un punteggio di 4. Queste sono definizioni che sono abbastanza banali e chiaramente non sono molto precise nella definizione di quello che è lo stato di validità del paziente, però sono molto importanti perché servono a permettere un dialogo tra i medici, servono a permettere a me che guardo una cartella clinica che è stata redatta da un mio collega a capire come era il paziente nella visita precedente per esempio o nelle visite di un anno fa. È chiaro che la valutazione soggettiva può essere molto più sofisticata, ma nello stesso tempo è molto meno riproducibile. Queste sono appunto delle scale che ci permettono di avere una riproducibilità di quella che è l'impressione del medico sullo stato di validità del paziente e questo è importante perché ha un valore prognostico, soprattutto nei linfomi diffusi a grandi cellule. Ha questo valore prognostico anche per altre malattie, ma nel caso del linfoma diffuso a grandi cellule è stato fatto un punteggio che è stato definito IPI (International Prognostic Index) che serve a dare una valutazione prognostica prima di iniziare il trattamento per i pazienti affetti da questo tipo di linfomi aggressivi e questo è stato fatto in maniera molto semplice, cioè cercando di dare dei parametri che possono essere raccolti da qualunque medico e in qualunque condizione. I parametri sono: − età, che ha un valore prognostico, dividendo i pazienti di età inferiore a 60 anni e superiore a 60 anni; − LDH che come vi dicevo è strettamente correlato a quella che è la capacità proliferativa della malattia linfomatosa; La β2-microglobulina ha la stessa importanza del LDH ( nelle slide del prof la β2microglobulina non viene riportata perché viene considerata una casistica americana e gli americani non fanno la β2-microglobulina o per lo meno solo in alcune situazioni. Gli autori che hanno stilato la casistica quindi non avevano a disposizione quel dato e non l'hanno inserito). − Performance Status, valutato secondo la scala ECOG, quindi 0, 1, 2, 3, 4; − Stadio di malattia, stadio iniziale 1 o 2, verso 3 o 4; − Interessamento extralinfonodale, cioè di strutture al di fuori di linfonodi fino a una sede o più di una sede. Molto semplicemente a questi 5 parametri è stato dato un punteggio 0 o 1 a seconda che il parametro fosse favorevole o sfavorevole e sulla base di queste semplici osservazioni sono state identificate diverse categorie di pazienti con diversa prognosi. Parliamo di pazienti tutti affetti da linfoma diffuso a grandi cellule e tutti trattati allo stesso modo, quindi uguale diagnosi istologica, uguale trattamento, ma i pazienti vanno differenziati soltanto per queste caratteristiche: età, LDH, Performance Status, Stadio e coinvolgimento extranodale. Vedete qui sono stati identificati 4 gruppi di rischio che sono: − basso rischio, se i pazienti avevano 0 o 1 di quei parametri positivi; − basso intermedio; − alto intermedio; − alto rischio, se i pazienti avevano da 4 a 5 di quei parametri positivi. (slide) Qui vedete la distribuzione dei casi che grossomodo è abbastanza omogenea e qui vedete la percentuale di remissioni complete. I pazienti a basso rischio hanno l'87% di probabilità di ottenere la scomparsa clinica della malattia, cioè la cosiddetta “remissione completa” contro la metà che si ottiene nei pazienti ad alto rischio. Ma soprattutto se voi vedete, la sopravvivenza a 5 anni è molto diversa, cioè i pazienti con basso rischio hanno una probabilità di guarigione (perché una sopravvivenza a 5 anni nel linfoma diffuso a grandi cellule significa guarigione) che è del 73%, quindi 3 su 4 guariscono. Viceversa i pazienti ad alto rischio hanno una probabilità di guarigione che è soltanto del 26%, cioè 1 su 4 guarisce. Capite bene dunque che c'è molta differenza tra queste due condizioni e ovviamente ci sono anche le condizioni intermedie. E questo basandoci soltanto su 5 parametri molto banali che probabilmente valgono per molte altre malattie, non solo per i linfomi non-Hodgkin perché vi ricordo che i 5 parametri sono l'età, LDH, il Performance Status, lo stadio e il coinvolgimento extranodale. Sono sicuro che se si mettono a fare una cosa del genere per il tumore della prostata cambiando probabilmente qualche cosa (invece di coinvolgimento extranodale mettono la localizzazione ossea di malattia) probabilmente ottengono lo stesso risultato, quindi sono parametri grossolani, ma già con questi parametri grossolani c'è una forte distinzione tra i pazienti, pur avendo questi pazienti lo stesso tipo istologico di malattia e pur avendo questi pazienti lo stesso tipo di trattamento. Quindi non è pensabile che noi identifichiamo una prognosi uguale per tutti i pazienti affetti da linfoma non-Hodgkin diffuso a grandi cellule. La prognosi dipende da tante caratteristiche. Già basta fare una distinzione così grossolana che già abbiamo una grande distinzione nella prognosi, figuriamoci poi quando ci vengono a dire che i linfomi sono tutti uguali!!! Qua stiamo parlando soltanto di linfomi diffusi a grandi cellule. Ma vi dicevo che queste sono delle distinzioni molto grossolane, adesso andiamo sempre di più verso la medicina molecolare e la medicina molecolare ci aiuta nella valutazione prognostica. Non è che la medicina molecolare si sostituisce a queste alterazioni abbastanza semplici e grossolane da poter identificare, ma sicuramente aggiunge qualcosa in più in maniera molto più costosa, in maniera molto più complicata, ma sicuramente aggiunge qualcosa in più. Uno dei parametri che si stanno affermando nella valutazione prognostica dei pazienti con linfoma è la tecnica dei microarrays che permette di valutare contemporaneamente e velocemente l'espressione di molti geni in un piccolo campione. (slide) Vedete qua sono dei piccoli vetrini con dei chips molto piccoli che sono poco più di 1cm e in questo cm ci stanno molti geni, fino a quarantamila geni tutti in una volta. Quindi capite bene come queste tecniche ci permettono di valutare bene una quantità molto elevata di geni perché una delle caratteristiche che dobbiamo valutare è proprio l'espressione di alcuni geni in questi pazienti. La “gene expression” può essere fatta valutando il DNA o mRNA o infine adesso si sta cercando di capire un po' meglio quella che è l'espressione proteica, la cosiddetta proteomica ( una fa parte della genomica, l'altra della proteomica). Quindi sempre di più vengono messe in atto queste tecniche che ci permettono di valutare in una piccolissima quantità di campione un numero elevatissimo di geni. Questi sono i principi su cui sono basati questi esami, questi arrays che sono basati fondamentalmente sulla base di sonde già predisposte che vanno a legarsi con il DNA o RNA del paziente e attraverso un sistema molto complesso si vengono a valutare in un chip un numero elevatissimo di geni. (slide) Questo è un esempio: ci sono dei quadratini e ad ogni quadratino corrisponde un gene e ad ogni linea orizzontale corrisponde un gene e ad ogni linea verticale corrisponde un paziente. La seconda riga è il gene della Lattico deidrogenasi che come vedete è acceso (il colore rosso significa che il gene è acceso, il colore blu che il gene è spento). Nei linfomi diffusi a grandi cellule il gene della LDH è quasi sempre acceso e questo conferma il fatto che questi pazienti hanno una elevata produzione di lattico deidrogenasi. Viceversa nei linfomi follicolari è spento. Ma vedete come esistono tutta una serie di geni che differenzia i linfomi follicolari dai linfomi diffusi a grandi cellule. Nei linfomi follicolari ad esempio sono accesi altri geni che invece sono spenti nei linfomi a grandi cellule. Ma limitando l'analisi ai linfomi a grandi cellule è stato osservato che esistono almeno tre tipi di linfomi a grandi cellule in termini di espressione del profilo genico e questi tre tipi sono: − i linfomi diffusi a grandi cellule che esprimono un profilo genico simile a quello del centro germinativo; − i linfomi diffusi a grandi cellule che esprimono un profilo genico come i linfociti attivati; − gruppo intermedio, un po' più piccolo. Questo ha permesso di fare una distinzione prognostica perché vedete come i pazienti che hanno un profilo di espressione genica come da centro germinativo vanno molto meglio rispetto a pazienti che hanno un profilo di espressione genica come da linfociti attivati. Questo permette di differenziare ulteriormente i pazienti in basso rischio e alto rischio e anche se prendete i pazienti che hanno un IPI, cioè quelle caratteristiche cliniche che vi ho fatto vedere per cui l'International Prognostic Index è considerato basso, se noi a questi pazienti facciamo una valutazione del profilo di espressione genica, vediamo che ulteriormente questo gruppo di pazienti viene suddiviso in : − un gruppo ad ottima prognosi che ha un profilo di espressione genica tipo linfociti del centro germinativo; − a peggiore prognosi che sono quelli che hanno un profilo di espressione genica tipo linfociti attivati. Questo nell'ambito dei pazienti che hanno un IPI basso, cioè che hanno un International Prognostic Index basso. Quindi questo ci dice che la valutazione che abbiamo fatto con l'International Prognostic Index ha una sua validità ed è molto importante perché permette di stratificare i pazienti in base al rischio prognostico, ma sei noi a questa valutazione grossolana, clinica aggiungiamo delle valutazioni di tipo biologico abbiamo ulteriori informazioni che ci permetto di ulteriormente classificare meglio questo tipo di pazienti. Questi esami ci hanno permesso di dividere i linfomi diffusi a grandi cellule in almeno due malattie che sono i pazienti che hanno i linfomi diffusi a grandi cellule che hanno un profilo di espressione genica tipo centro germinativo e un altro tipo che ha un profilo di espressione genica tipo cellule attivate. Probabilmente l'ultimo tipo deriva da una zona subito al di fuori del centro germinativo, alla periferia del centro germinativo e hanno la caratteristica di avere una attivazione costitutiva di NFkB che è una molecola che ha un ruolo molto importante nella proliferazione della cellula. Se ricordate ne avevamo parlato a proposito del linfoma di Hodgkin quando abbiamo visto come NFkB può essere una molecola stimolata dal virus di Epstein-Barr e questo potrebbe essere uno dei motivi per cui il virus di Epstein-Barr determini una trasformazione in senso oncologico dei linfociti colpiti dal virus di Epstein-Barr. Viceversa nei pazienti che hanno un linfoma diffuso a grandi cellule di tipo “germinal centre”, in questo caso è molto più frequente la stessa traslocazione che abbiamo visto per i linfomi follicolari, la (14;18), condizione questa che determina una iperespressione del bcl-2. Questi pazienti abbiamo visto hanno un prognosi più favorevole, mentre questi altri pazienti hanno una prognosi peggiore. Tutto questo è importante perché ad esempio adesso ci sono dei farmaci che sono rivolti ad inibire l'attività di NFkB e ci sono dei farmaci rivolti ad inibire bcl-2. Ancora sono farmaci in fase sperimentale ma si è abbastanza sicuri che questi farmaci sono dei farmaci attivi e che hanno un effetto su questo tipo di malattia. Però è chiaro che bisogna indirizzare questo tipo di terapia sulla base delle caratteristiche biologiche del paziente, cioè non si possono trattare con gli inibitori di NFkB tutti i pazienti con il linfoma diffuso a grandi cellule, ma bisognerà trattare soltanto quei pazienti che hanno una attivazione costitutiva di NFkB, oppure bisogna trattare con inibitori di bcl-2 soltanto questo gruppo di pazienti che hanno l'iperespressione di bcl-2. Quindi capite bene che questi aspetti biologici che informano praticamente tutti i linfomi sono aspetti biologici particolarmente sofisticati, devono essere ricercati il più possibile, noi ancora non abbiamo una tecnologia a basso costo che ci permetta di evidenziare questo tipo di effetti. Considerate che fare un microarray andando a guardare un po' tutto il genoma umano grossomodo costa mille euro a campione!! Quindi sono delle spese ancora eccessive per cui non è entrata nella pratica clinica questo tipo di informazione, ma sicuramente è entrata nella pratica clinica tutta una serie di valutazioni di biologia molecolare un pochettino più ristrette ma che ci possono essere di ausilio. Proprio perché queste cose sono particolarmente costose si è cercato di trovare un escamotage, cioè andare a riprodurre con l'immunoistochimica quelle informazioni che ci sono state dalla biologia molecolare. E sicuramente l'immunoistochimica in parte ci aiuta perché per esempio andando a marcare le cellule con il bcl-6 o con il CD10 prendiamo la maggior parte delle cellule del centro germinativo. Andando a marcare le cellule con il CD138 o con il MUM1 prendiamo le cellule post-centro germinativo. Qualche anno fa è stata proposta questa distinzione per cui in pratica andando a giocare un po' con questa immunoistochimica ci permette di differenziare i linfomi diffusi a grandi cellule che sono di tipo centro germinativo rispetto a tutto il resto. Questo non è del tutto vero nel senso che l'immunoistochimica non riesce a riprodurre in maniera fedele quella che è l'espressione del “gene expression profiling”. Il “gene expression profiling” è stato anche adottato nello studio del linfoma follicolare e qui è spuntata fuori una cosa interessante. È spuntato fuori che in realtà c'è una differenza in termini prognostici tra i linfomi follicolari che hanno un profilo di espressione genica che interessa più che altro la risposta immunologica al linfoma e che sono stati differenziati in risposta di tipo 1 e risposta di tipo 2 a seconda dei geni che sono coinvolti in questa risposta immunologica e la cosa interessante è che i pazienti che avevano anche in immunoistochimica una quantità di macrofagi superiore vanno peggio rispetto ai pazienti che non la avevano. (slide) Vedete questa è una sezione di un linfonodo, questi punti neri sono la colorazione per i macrofagi, quindi cellule non neoplastiche ma cellule che in qualche modo fanno parte del sistema immunitario del paziente. Stranamente quello che si è visto è che pazienti con basso numero di macrofagi vanno molto meglio rispetto ai pazienti con alto numero di macrofagi. Quello che uno si aspetta è che una reazione immunologica del paziente rispetto alla malattia sia più efficace nel combattere la malattia rispetto a chi non ha questa reazione immunologica. In realtà per il linfoma follicolare sembra esattamente il contrario e quindi sono state fatte delle ipotesi dove i pazienti che hanno tante cellule di linfoma follicolare immerse in tante cellule T, ma pochi macrofagi vanno molto meglio rispetto ai pazienti che hanno molti macrofagi. Al momento non c'è una spiegazione per questo tipo di osservazione. Tutto questo per dirvi come si evolve continuamente la ricerca e le informazioni che noi abbiamo su quella che è la linfomagenesi e la prognosi e le caratteristiche biologiche e cliniche di questi pazienti per cui mentre prima i markers prognostici erano basati sugli aspetti clinici del paziente, poi si è passati all'immunoistochimica (vedere per esempio l'espressione di bcl-2), adesso confidiamo su quelle che sono le alterazioni genetiche non solo del tumore, ma anche su quelli che sono gli aspetti peculiari dell'ospite perché per la risposta al trattamento non vale soltanto sapere quanto aggressivo è il tumore, quanto il tumore ha delle alterazioni molecolari o genetiche in generale, ma è importante anche il fatto che il paziente viene trattato con una determinata chemioterapia e questa chemioterapia può avere diversi effetti. Ci sono pazienti che hanno poca efficacia dalla chemioterapia ma tanti effetti collaterali, alcuni pazienti hanno il contrario e questo dipende dal polimorfismo del paziente, dalla capacità del paziente di metabolizzare questi farmaci, che è un fatto squisitamente individuale. Ognuno di noi ha dei polimorfismi che rendono più o meno attiva la terapia e li vedremo successivamente. - Possiamo dare qualche accenno anche per qualche altro linfoma particolarmente importante, per esempio il linfoma a grandi cellule anaplastiche caratterizzato dalla positività per il CD30 e da una traslocazione specifica (2;5). È un linfoma particolarmente aggressivo, ma anche particolarmente sensibile al trattamento per cui è uno di quei linfomi che se lasciato da solo è assolutamente mortale risponde molto bene al trattamento. - Un altro linfoma che ha queste caratteristiche è il linfoma di Burkitt. Il linfoma di Burkitt è un linfoma molto aggressivo, caratterizzato da una associazione con il virus di Epstein-Barr, è endemico in Africa, è presente però anche dalle nostre parti dove spesso dà delle localizzazioni extralinfonodali e in particolare è tipica la localizzazione a livello dell'ovaio e dell'intestino. Il linfoma di Burkitt è caratterizzato da una traslocazione (8:14) o comunque da un interessamento del cromosoma 8 che viene appunto ad essere interessato in ogni traslocazione del linfoma di Burkitt e questo perché nel cromosoma 8 ci sta il gene c-myc. Il gene c-myc è un gene che serve alla proliferazione cellulare. Quando per effetto della traslocazione questo gene viene sottoposto all'azione di un enhancer come nel caso specifico dei geni delle immunoglobuline che stanno sul cromosoma 14, in questo caso il c-myc sta sempre acceso e determina una proliferazione continua del tessuto linfomatoso. Tipicamente è un linfoma che interessa la mandibola in Africa. La zona dell'Africa in cui è endemico il linfoma di Burkitt è la stessa zona in cui è endemica la malaria e quindi si pensa che la combinazione del virus di Epstein-Barr più il plasmodio della malaria abbia un ruolo patogenetico nello sviluppo di questo tipo di linfoma. - Un'altra entità nosografica importante è il cosiddetto linfoma mantellare chiamato così perché appunto interessa il mantello del centro germinativo. Questo è un linfoma che si caratterizza per il fatto di avere una iperespressione di bcl-1 e della cosiddetta ciclina D1, un marcatore molto specifico del linfoma mantellare. La caratteristica di questo linfoma mantellare è che morfologicamente è un linfoma che assomiglia molto ai linfomi indolenti, prima veniva addirittura classificato tra i linfomi indolenti, però è un linfoma che ha delle caratteristiche cliniche peculiari e soprattutto è un linfoma che risponde molto poco alla terapia per cui se su un piano morfologico è un linfoma classificabile come indolente, su un piano clinico è un linfoma sicuramente aggressivo perché è un linfoma che deve essere trattato con una chemioterapia la più aggressiva possibile. È caratterizzato da una traslocazione (11:14), un “pezzo” di 14 va a finire sul cromosoma 11 in cui ci sta la ciclina D1 che quindi viene iperespressa. È un linfoma usualmente abbastanza diffuso, è presente in età avanzata, dà la cosiddetta poliposi linfomatoide. La poliposi linfomatoide non è altro che una localizzazione intestinale del linfoma mantellare che dà molti polipi intestinali. La localizzazione intestinale con questi polipi è proprio tipica del linfoma mantellare. La terapia del linfoma mantellare è una terapia particolarmente aggressiva e almeno nei pazienti giovani dobbiamo fare una procedura trapiantologica per ottenere una buona risposta. - Dei linfomi a cellule T non ne parliamo, nel senso che sono dei linfomi molto meno frequenti rispetto ai linfomi a cellule B e ripercorrono un po' la stessa distinzione dei linfomi a cellule B, cioè ci sono i linfomi indolenti e ci sono i linfomi aggressivi. Usualmente i linfomi della cute per esempio sono linfomi a cellule T e questi però sono generalmente abbastanza indolenti, ma poco sensibili al trattamento. In generale i linfomi a cellule T sono poco sensibili al trattamento per cui se da un lato in qualche modo ripercorrono lo stesso grado di malignità dei linfomi a cellule B (più indolenti o più aggressivi) però in generale sono tutti meno responsivi al trattamento rispetto ai linfomi a cellule B per cui avere un linfoma a cellule T di tipo aggressivo è molto peggio rispetto ad avere un linfoma a cellule B aggressivo perché quello B aggressivo può rispondere bene al trattamento, mentre quello T aggressivo non risponde al trattamento. LINFOMI MALT Infine abbiamo i linfomi cosiddetti MALT. I linfomi MALT sono i linfomi che interessano il tessuto linfoide associato alla mucosa. MALT è un acronimo che sta per Mucose-Associated Lymphoid Tissue. I linfomi MALT sono prevalentemente linfomi del tubo gastroenterico, ma possono colpire anche tutte le ghiandole, possono colpire le ghiandole salivari, possono colpire le ghiandole lacrimali, possono colpire anche il polmone, possono colpire tutte le zone dove ci sono ghiandole. Però il prototipo del linfoma MALT è il linfoma intestinale e in particolare il linfoma gastrico. Il tessuto MALT è presente in certi siti extranodali in condizioni normali, per esempio le placche di Peyer, lì c'è del tessuto linfoide, mentre nello stomaco in condizioni normali non c'è tessuto linfoide. Il tessuto linfoide c'è quando è presente una infiammazione dello stomaco oppure ad esempio della tiroide e questa è la conditio sine qua non per sviluppare il linfoma. In questi tessuti prima devono arrivare i linfociti e poi i linfociti li trasformano e in particolare questa trasformazione è un po' difficile da evidenziare su un piano anatomo-patologico, su un piano istologico per cui molto spesso esiste una difficoltà nel fare una diagnosi differenziale tra una gastrite e un linfoma e ci sono appunto dei criteri anatomo-patologici che sono utili per questa distinzione. Fondamentalmente quello che abbiamo scoperto negli ultimi anni è che comunque c'è un'evoluzione dalla gastrite al linfoma, cioè il linfoma MALT dello stomaco non può svilupparsi senza aver avuto prima il paziente la gastrite e la gastrite è più spesso una gastrite indotta da Helicobacter pylori che molto spesso precede di anni lo sviluppo del linfoma. Quello che è stato un pochettino accertato è che esiste questa gastrite cronica da Helicobacter pylori, la quale gastrite cronica determina una attivazione di T linfociti, i quali determinano una stimolazione per contatto dei linfociti B. Siccome c'è la gastrite ci sono anche tanti neutrofili e voi sapete che i neutrofili liberano dei radicali liberi, in particolare dei superossidi e questi radicali liberi hanno probabilmente un effetto di indurre delle alterazioni geniche su questi B linfociti che sono in attiva proliferazione. Tutto ciò fa sì che ci sia una trasformazione, questi linfociti acquisiscono alterazioni genomiche che li fanno trasformare e li fanno diventare cellule linfomatose. Le cellule linfomatose per la loro crescita, almeno nella fase iniziale, hanno sempre bisogno di Helicobacter. Successivamente in una fase più tardiva diventano indipendenti da Helicobacter. Quindi si ha infezione da Helicobacter, si acquisisce tessuto MALT, ci possono essere delle alterazioni genomiche tipo la traslocazione (1;14) o altro tipo di traslocazione, la trisomia del cromosoma 3, del 12, del 18 e tutto questo induce la formazione di un linfoma MALT. In questa fase il linfoma MALT è ancora Helicobacter pylori-dipendente. Successivamente poi per aggiunta di altre alterazioni genomiche si sgancia da questa dipendenza e diventa un linfoma anche trasformato o con delle altre alterazioni come (11:18) che non lo rendono più dipendente da Helicobacter. Tutto questo è molto importante perché ha fatto sì che noi possiamo trattare i linfomi gastrici in questa fase soltanto togliendo l'Helicobacter e questa vedete è stata una rivoluzione nel trattamento dei linfomi gastrici! Fino a quando il prof era un giovane ematologo i linfomi gastrici venivano trattati con la gastrectomia totale cioè se un paziente aveva un linfoma MALT veniva mandato da un chirurgo il quale gli toglieva lo stomaco. L'avere scoperto che esistono queste correlazioni tra lo sviluppo del MALT e l'Helicobacter pylori e l'aver scoperto che in una fase iniziale il linfoma MALT dipende ancora da Helicobacter ha fatto sì che alcuni pazienti sono stati trattati con una terapia anti-Helicobacter e hanno ottenuto la remissione completa del linfoma. Quindi è bastato trattare questi pazienti con una terapia antibiotica per risparmiargli una gastrectomia, il che non è poco. Qual è la clinica del linfoma gastrico? Usualmente il paziente va dal medico perché ha un dolore epigastrico, dispepsia, nausea e vomito, talvolta ha l'ulcera perforata, talvolta ha anche le emorragie e ha un reperto endoscopico abbastanza variabile. Talvolta c'è solo un ispessimento delle pliche perché il linfoma si propaga, cresce soprattutto nella sottomucosa. Quindi non è come il carcinoma che ha una proliferazione esofitica per cui c'è proprio un cavolfiore che cresce dentro lo stomaco ma c'è una proliferazione sottomucosa del linfoma che spesso dà dei segni endoscopici non così evidenti. Colpisce generalmente l'antro o il corpo, cioè dove c'è la più alta concentrazione di Helicobacter ed è un linfoma che tende a rimanere localizzato allo stomaco. Come si fa la stadiazione del linfoma? Bisogna fare ovviamente la gastroscopia, ma bisogna fare soprattutto adesso la cosiddetta ecoendoscopia, cioè una ecografia in corso di endoscopia e questo ci permette di vedere lo spessore della parete gastrica, cioè di quanto la parete gastrica è stata coinvolta dal processo linfomatoso, se è stata coinvolta soltanto la sottomucosa oppure se è stata coinvolta anche la muscolaris mucosae o tutto lo spessore della parete dello stomaco perché questo fa la differenza. Ovviamente bisogna andare a cercare l'Helicobacter e fare questa famosa ecoendoscopia. Nelle prime fasi, quando la malattia non interessa tutto lo stomaco, ma quando la malattia ancora è limitata alla sottomucosa per esempio e quindi significa che la malattia è ancora nella sua prima fase, questo significa che la malattia è Helicobacter pylori-dipendente, quindi questi sono i pazienti che vanno trattati con la sola terapia antibiotica. Quando parliamo di terapia dei linfomi gastrici dobbiamo intanto considerare che i MALTomi gastrici sono quelli che vanno meglio di tutti rispetto a tutti gli altri linfomi a localizzazione extranodale, questo a prescindere dalla terapia che facciamo. Possiamo utilizzare comunque diverse opzioni terapeutiche per questi linfomi gastrici. La prima opzione terapeutica è la terapia eradicante, cioè eradicante l'Helicobacter. Oppure si può fare la resezione chirurgica che si faceva in passato oppure si può fare la radioterapia, la chemioterapia e gli anticorpi monoclonali. Il prof mostra esempio della terapia antibiotica utilizzata nella terapia dei linfomi gastrici e la risposta agli antibiotici dei linfomi gastrici allo stadio 1, cioè quelli confinati alla sottomucosa. Come vedete, nei vari studi che sono stati fatti, la percentuale di risposte complete è estremamente elevata, va almeno dal 56% al 100% dei pazienti con MALT gastrico in stadio iniziale. Ci sono quelli che non rispondono alla terapia anti-Helicobacter anche se hanno uno stadio iniziale e sono quelli che hanno la traslocazione (11:18). Non sappiamo bene per quale motivo, ma i pazienti che hanno la traslocazione (11:18) per la maggior parte, circa il 67%, sono resistenti all'eradicazione di Helicobacter. Però mentre alcuni casi di linfoma dello stomaco, che è un linfoma MALT e quindi un linfoma a basso grado di malignità, possono evolvere verso un linfoma diffuso a grandi cellule e quindi un linfoma ad alto grado di malignità, quelli che hanno la traslocazione (11:18) non evolvono mai in linfoma ad alto grado di malignità. Quindi non rispondono bene alla terapia anti-Helicobacter, ma neanche evolvono verso il linfoma ad alto grado. Cosa succede se un paziente non risponde al trattamento anti-Helicobacter? In questo caso ognuno si comporta in maniera un po' differente. Ci sono le strutture ematologiche che preferiscono ovviamente non fare la gastrectomia ma ci sono ancora alcuni centro oncologici soprattutto chirurgici che preferiscono fare la gastrectomia. Direi che la gastrectomia è ormai quasi del tutto abbandonata. (slide) Questa è una casistica in cui sono state valutate retrospettivamente un approccio chirurgico rispetto ad un approccio conservativo di chemioterapia o di trattamento anti-Helicobacter e vedete che non c'è nessuna differenza in termini di sopravvivenza e anzi esiste una pubblicazione che dice che in tutti i pazienti con linfoma gastrico anche aggressivo quelli trattati con chemioterapia o con chirurgia più chemioterapia vanno meglio rispetto a quelli trattati con la chirurgia soltanto, quindi quello che è importante è trattare i pazienti con un approccio di tipo medico e non con un approccio di tipo chirurgico. Nei pazienti che non rispondono si può fare anche l'anticorpo monoclonale che si chiama Rituximab, che è un anticorpo monoclonale anti-CD20. Questo dà la possibilità di parlarvi di quello che è l'impiego degli anticorpi monoclonali in ematologia e in particolare nei linfomi. (slide) Qui vedete rappresentata l'ontogenesi del linfocita B e a seconda della tappa dell'ontogenesi in cui si ha l'alterazione possono svilupparsi diverse malattie. Stem cell Pre-B Plasma cell Early B intermediate B Mature B Plasmacytoid B Per cui ad esempio i pazienti che hanno una trasformazione neoplastica del linfocita pre-B sono pazienti che avranno una leucemia acuta. Viceversa i pazienti che avranno una trasformazione neoplastica del linfocita B maturo sono pazienti che avranno una malattia linfomatosa. Questi linfociti hanno una espressione antigenica differente e se vedete qui il CD20 è una molecola peculiare perché non è presente nei pre-B linfociti e non è presente neanche nei linfociti più maturi che sono le plasmacellule, ma è presente soltanto in queste tappe intermedie dell'ontogenesi, della maturazione dei linfociti B. Numerose patologie linfoproliferative hanno una elevata espressione di CD20: − la cosiddetta “tricoleucemia” di cui parleremo, − i linfomi a grandi cellule, − il linfoma di Burkitt, − il linfoma della zona marginale, − i linfomi follicolari ecc.. . Nei confronti di quest'antigene è stato prodotto un anticorpo monoclonale che si chiama Rituximab che ha rappresentato un notevole passo avanti in termini di efficacia della terapia che abbiamo contro questo tipo di linfomi. (slide) Questa per esempio è l'esperienza della British Columbia pre e post Rituximab nei linfomi diffusi a grandi cellule e come vedete c'è stato un netto guadagno nell'aggiunta del Rituximab nella chemioterapia sia per i pazienti di età superiore ai 60 anni, sia per i pazienti di età inferiore ai 60 anni. Lo stesso vale per i linfomi follicolari, qui vedete la combinazione di una chemioterapia che si chiama CVP (Ciclofosfamide Vincristina Prednisone) verso la stessa terapia con l'aggiunta del Rituximab e vedete come la sopravvivenza è nettamente migliorata rispetto ai pazienti che venivano trattati solamente con chemioterapia. Lo stesso vale sempre per questa casistica di linfomi a basso grado di malignità e tutto sommato il Rituximab è un farmaco gravato da pochi effetti collaterali. Gli unici effetti collaterali a distanza sono una certa riduzione della conta delle cellule B e della produzione di immunoglobuline. Devo dire però che stanno cominciando adesso ad esserci dei warnings un pochettino nell'utilizzo di questi farmaci perché questo farmaco, l'anti-CD20, che è estremamente efficace nei linfomi, è stato un po' utilizzato anche per tante altre malattie dove si voleva abbassare un pochettino la quota di produzione di immunoglobuline e questo è stato utilizzato in molte malattie autoimmuni, lo utilizziamo ad esempio nelle piastrinopenie autoimmuni, nelle anemie emolitiche autoimmuni, ma è stato utilizzato anche in malattie reumatologiche e purtroppo però sono stati segnalati dei casi di leucoencefalopatia multifocale progressiva in pazienti trattati con Rituximab come se un abbassamento dei poteri di difesa, un abbassamento della capacità di produrre immunoglobuline possa aver favorito un'infezione dovuta a prioni che determinano la leucoencefalopatie multifocale progressiva, in poche parole il morbo della mucca pazza. Tutto questo è stato collegato all'impiego prolungato di farmaci che comunque hanno come effetto l'inibizione di una parte di sistema immunitario. Per fortuna non è stata fatta nessuna segnalazione in ematologia di questo tipo di infezione però c'è la possibilità che altre infezioni siano aumentate in pazienti trattati con Rituximab. Certamente ancora abbiamo molto da migliorare in termini di impiego di questi farmaci, di tutti gli anticorpi monoclonali ma in particolare del Rituximab, ma soprattutto per migliorare questi effetti dobbiamo conoscere meglio come funzionano questi farmaci. E allora volevo un po' entrare nel particolare di come funziona il Rituximab che poi è un po' un modo in cui funzionano molti anticorpi monoclonali. Quando noi utilizziamo un anticorpo monoclonale, in particolar modo il Rituximab, questo può avere diversi effetti. Un primo effetto è quello del blocco della proliferazione. (slide) Considerate che questa è la cellula bersaglio del Rituximab e questo anticorpo in verde è il Rituximab. Il Rituximab può da un lato bloccare la proliferazione della cellula, dall'altro indurre direttamente l'apoptosi della cellula, cioè l'anticorpo si lega al suo antigene e questo con un meccanismo piuttosto complesso che si chiama lipid raft determina una sorta di polarizzazione delle glicoproteine della membrana lipidica e quindi determina poi l'apoptosi della cellula. Ma un altro aspetto molto più importante rispetto a quello dell'apoptosi indotta è l'apoptosi complemento-mediata, cioè questo anticorpo si lega al suo antigene e per effetto del complemento induce una lisi della cellula. Infine un altro aspetto molto importante è la cosiddetta ADCC cioè Citotossicità Anticorpomediata dove in pratica l'anticorpo monoclonale fa da ponte tra le cellule bersaglio e le cellule effettrici che sono gli NK, i monociti, i polimorfonucleati del paziente, quindi qui c'è una connessione indotta dall'anticorpo monoclonale tra le cellule effettrici e le cellule bersaglio e questa connessione avviene perché l'anticorpo monoclonale si va a legare su un recettore di queste cellule effettrici che si chiama FcγR (R sta per Receptor), di cui esistono diversi tipi, FcγRI, FcγRII, FcγRIII, ma tutti quanti sono governati da alcuni polimorfismi che tutti noi abbiamo. (slide) In particolare FcγRII e FcγRIII possono presentare degli amminoacidi in posizione differente per cui in particolare vedete rappresentato FcγRIIIa che può essere omozigote per valina o può essere eterozigote oppure omozigote per fenilalanina. Se il soggetto ha la fenilalanina, questo configura un ingombro sterico che impedisce all'anticorpo monoclonale di legarsi in maniera stretta con questo recettore, quindi il legame è un pochettino più blando e quindi meno efficace. Tutto questo fa parte del polimorfismo individuale e ritorniamo al discorso di prima, ossia l'importanza del polimorfismo nella prognosi finale del paziente. Se questo legame tra la cellula tumorale e le cellule effettrici indotto dall'anticorpo monoclonale è un legame debole, la ADCC sarà inefficace, se è un legame forte la ADCC sarà efficace. Il legame forte sarà soltanto per la valina, il legame debole per la fenilalanina. Quindi se un paziente è omozigote per valina avrà un legame forte, se un paziente è omozigote o eterozigote per fenilalanina avrà un legame debole. È stato dimostrato appunto nei pazienti trattati con solo Rituximab che vanno molto meglio i pazienti che sono omozigoti per valina rispetto agli altri. Il problema però è che studiando ampiamente questo aspetto sia nella nostra casistica che in altre casistiche purtroppo gli omozigoti per valina costituiscono una minoranza dei pazienti e quando noi andiamo ad utilizzare la combinazione chemioterapico più anticorpo monoclonale in realtà annulliamo questo effetto del polimorfismo genetico. In questo senso si stanno sviluppando degli anticorpi che hanno una maggiore affinità per il recettore Fcγ e questo probabilmente può permettere di superare anche questo aspetto. (slide) Ma vedete qui quanto la scienza sta cercando di fare nel produrre tanti nuovi anticorpi monoclonali nei confronti delle cellule dei linfomi a cellule B. Contro il CD20, il famoso Rituximab, quello presente in commercio e che noi utilizziamo, me tutta una serie di altri anticorpi monoclonali sempre diretti contro il CD20 che hanno delle caratteristiche di maggiore affinità per il CD20. Adesso c'è un anticorpo monoclonale contro il CD22, contro il CD23, contro il CD40, è in commercio un anticorpo monoclonale contro il CD52 e tutta una serie di anticorpi monoclonali compreso l'anticorpo monoclonale anti-VEGF che sarebbe il fattore di crescita dell'angiogenesi che è utile in molte neoplasie. Un altro anticorpo estremamente interessante è il Bait (?), un anticorpo bispecifico e in pratica ha da un lato un anti-CD3 e quindi si lega alle cellule T, dall'altro un anti-CD19 e quindi si lega alle cellule linfomatose e in pratica questo permette di legare le cellule T alle cellule tumorali, condizione questa che non avviene in natura perché in natura per legare le cellule T alle cellule tumorali c'è bisogno di tutta una serie di co-stimolatori che per qualche motivo (perché la cellula tumorale nasconde alcuni antigeni o perché c'è una diversa classe MH2 ecc..) per qualche motivo non avviene. Questo anticorpo costringe questo contatto e questo però è ancora in fase molto sperimentale e abbiamo bisogno di altre informazioni. L'Epratuzumab è un anticorpo anti-CD22 che sta dimostrandosi efficace in numerosi linfomi resistenti al Rituximab e ci sono numerosi studi che dimostrano appunto l'efficacia dell'Epratuzumab aggiunto al Rituximab e alla chemioterapia nell'indurre una buona risposta in questi pazienti. Infine c'è un altro aspetto degli anticorpi monoclonali da utilizzare, il fatto che questi anticorpi possono anche veicolare una immunotossina. Questo è importante perché ad esempio questo è un anticorpo anti-CD22 (slide) che legato ad una antraciclina può veicolare questo farmaco direttamente nelle cellule CD22-positive, cioè nelle cellule linfomatose. Ma non soltanto si possono veicolare farmaci, si possono veicolare anche sostanze radioattive. (slide) Questo è un altro anticorpo anti-CD20 a cui è legato un radionuclide, in particolare l'Ittrio 90 che permette di fare non solo una immunoterapia ma una radioimmunoterapia. L'anticorpo si lega all'antigene CD20, ma ci sono anche cellule che non hanno il CD20 oppure in cui il CD20 è nascosto, oppure dove il farmaco non arriva per via del fatto che ci sono tante cellule vicine e lì il farmaco non ci arriva. Tutto questo è superato dal cosiddetto effetto “cross-fire”, cioè veicolando questi anticorpi monoclonali un radionuclide, permettono di fare una sorta di radioterapia locale, dove questo anticorpo monoclonale è stato veicolato e quindi questo permette di indurre la distruzione delle cellule con la radioterapia in un arco che è stato calcolato essere di qualche millimetro. Infine un altro anticorpo monoclonale utilizzato in ematologia è l'anti-CD52 (MabCampath). Il CD52 è un antigene espresso su tutti i linfociti, monociti, spermatozoi, sugli eosinofili, non è espresso sulle cellule emopoietiche e questo anti-CD52 è stato utilizzato contro la leucemia linfatica cronica e per i linfomi. (slide) Vedete qui una biopsia osteo-midollare di un paziente pre-Campath che è il nome di questo anti-CD52 e post-Campath, dove vedete che c'è una notevole clearance di tutte queste cellule neoplastiche e quindi l'infiltrazione leucemica è fortemente ridotta. Però questo farmaco ha numerosi effetti collaterali che si verificano soprattutto al momento della iniezione del farmaco come brividi, febbre, nausea, vomito, ipotensione ecc.. che però si riducono mano a mano che si va avanti con la somministrazione, ma la cosa importante è che c'è una forte deplezione dei linfociti T in pazienti trattati con il Campath. (slide) Questi sono i linfociti T dei pazienti trattati con Campath e vedete come in tutti i pazienti si ottiene una forte deplezione linfocitaria che perdura per diversi mesi. Tutto questo fa sì che i pazienti siano particolarmente esposti alle infezioni, in particolare ad infezioni da Cytomegalovirus che rimane una delle infezioni più importanti nei pazienti trattati con questo tipo di terapia, per cui è necessario in questi pazienti non solo fare una profilassi antivirale, ma fare spesso una sorveglianza delle infezioni da CMV. Siamo costretti a richiedere l'antigene … (01:16:13) per CMV quasi ogni settimana nei pazienti che fanno questo tipo di terapia. Al di là del CMV sono pazienti che hanno una profonda depressione del sistema immunitario, che determina un rischio estremamente elevato di infezione. Lezione 9 Il prof mostra le risposte esatte al test sui linfomi Hodgkin. 1) Da quali cellule derivano le cellule di Reed-Sternberg? Dai linfociti B. 2) Quanti sono i sottotipi istologici del linfoma di Hodgkin classico? Sono 4. I 4 sottotipi corrispondono a: - a prevalenza linfocitaria; - sclerosi nodulare; - cellularità mista; - deplezione linfocitaria. Questo dipende dal grado di presenza di cellule di Sternberg e di cellule accessorie. 3) Per quale virus è ipotizzato un ruolo patogenetico nel linfoma di Hodgkin? Il virus di Epstein-Barr (anche altri virus però sono stati coinvolti). 4) Qual è la via più importante di propagazione del linfoma di Hodgkin? Per contiguità. 5) Quali sono i sintomi che definiscono la malattia B? Febbre, perdita di peso e sudorazione notturna. Vi ricordo che il prurito fa parte della possibile sintomatologia dell'Hodgkin, ma non viene considerato sintomo B, quindi quelli considerati sintomi B sono: febbre, perdita di peso e sudorazione notturna. 6) Quanti sono i possibili stadi clinici nel linfoma di Hodgkin? I possibili stadi clinici sono 4: − I Stadio: una singola sede linfonodale; − II Stadio: due o più sedi linfonodali dallo stesso lato del diaframma; − III Stadio: due o più sedi da ambedue i lati del diaframma; − IV Stadio: malattia extralinfonodale. 7) Quale indagine radiologica si può considerare obsoleta nel linfoma di Hodgkin? La linfografia. Ormai non si usa più, forse lo troverete in qualche vecchio libro, ma non si usa più. 8) Quali di questi parametri non ha importanza prognostica nel linfoma di Hodgkin? La piastrinopenia. Invece localizzazione mediastinica, l'età e il sesso hanno importanza prognostica nell'Hodgkin. 9) Il trattamento degli stadi iniziali prevede l'impiego: Negli stadi iniziali utilizziamo poca chemioterapia e pochissima radioterapia, però le utilizziamo ambedue. Mentre prima si utilizzava per esempio soltanto la radioterapia per campi estesi, adesso la radioterapia si utilizza ancora con dei campi estremamente piccoli, in pratica soltanto nella sede della neoplasia Per quanto riguarda i quiz dei linfomi non-Hodgkin. 1) lncidenza dei linfomi non-Hodgkin è più elevata: negli anziani. 2) Le alterazioni cromosomiche nei linfomi non-Hodgkin coinvolgono più frequentemente: Il cromosoma 14. 3) La proteina bcl-2 è più frequentemente espressa nei linfomi: Follicolari. Devo dire che anche in alcuni linfomi a grandi cellule è espressa la bcl-2, però tipicamente i follicolari sono quelli in cui c'è una maggiore espressione della bcl-2. 4) I linfomi follicolari si presentano più frequentemente in stadio: Avanzato. Non perché i linfomi follicolari siano più aggressivi o perché siano in qualche modo con maggiore quantità di malattia, ma siccome più spesso danno luogo a localizzazioni extralinfonodali, questo li fa passare automaticamente di stadio. Anche se hanno una malattia piccola, però frequentemente danno una localizzazione osteomidollare per esempio, quindi per definizione diventano stadio IV, ma questo non vuol dire che abbiano tanta malattia. Magari hanno soltanto la localizzazione osteo-midollare e un piccolo linfonodo interessato, quindi sono già per definizione stadio IV anche se in realtà il carico di malattia, il carico tumorale può anche essere più basso. Al di là di questo, siccome sono malattie che insorgono in maniera piuttosto indolente, crescono in maniera piuttosto indolente, molto spesso la diagnosi è abbastanza tardiva proprio perché il paziente non si accorge di questa malattia, quindi troviamo magari pazienti che hanno una malattia disseminata, in uno stadio avanzato, proprio perché è una malattia indolente. 5) Il Rituximab è un anticorpo: Anti-CD20. 6) L'IPI score è basato sui seguenti parametri: Età, LDH, Performance Status (PS), Stadio e siti extranodali. 7) Rispetto ai linfomi MALT dello stomaco l'infezione da Helicobacter pylori è: Antecedente. Il linfoma MALT è sempre preceduto da un infezione da Helicobacter pylori che poi si trasforma in un processo neoplastico, ma spesso ancora dipendente da Helicobacter, quindi basta togliere Helicobacter con una terapia antibiotica perché regredisce anche la malattia linfomatosa. 8) Oltre allo stomaco i linfomi MALT possono colpire: Sia le ghiandole salivari che i polmoni, che gli annessi oculari. MALT significa Tessuto Linfoide Associato alle Mucose, quindi dove c'è una mucosa e dove c'è uno stato di infezione, di infiammazione della mucosa perché arrivano i linfociti, lì ci può essere poi una trasformazione in senso linfomatoso. 9) Sul piano clinico i linfomi mantellari sono considerati: Linfomi aggressivi. Perché sebbene il linfoma mantellare sia un linfoma che sul piano morfologico è considerato un linfoma indolente, poi in realtà sul piano clinico è un linfoma aggressivo perché è un linfoma particolarmente resistente al trattamento, quindi anche se poi non ha una velocità di crescita molto rapida e anche se ha un aspetto morfologico di tipo indolente, tuttavia poi clinicamente diventa aggressivo perché risponde poco al trattamento. LEUCEMIA LINFATICA CRONICA Oggi parleremo di un'altra malattia linfoproliferativa che è la leucemia linfatica cronica, “parente” stretta dei linfomi, soprattutto dei linfomi a basso grado di malignità, solo che si presenta con una presentazione leucemica, cioè soprattutto nel sangue periferico, ma in realtà è un processo linfoproliferativo. Questa malattia è molto importante perché è la leucemia più comune. La leucemia linfatica cronica, almeno nei Paesi occidentali, è la leucemia più comune. È molto meno frequente in Asia e nei Paesi orientali in generale, non sappiamo bene per quale motivo, probabilmente ci sarà una motivazione legata alla razza. Tipicamente interviene in persone anziane ed è più comune negli uomini rispetto alle donne. Come si fa la diagnosi di leucemia linfatica cronica? Si tratta di pazienti che vengono perché hanno un emocromo dove c'è un aumento dei globuli bianchi e questo aumento dei globuli bianchi è sostenuto dai linfociti. Quindi c'è un aumento dei globuli bianchi e la formula leucocitaria è invertita, nel senso che ci sono più linfociti che neutrofili. Abbiamo fondamentalmente una linfocitosi, che almeno nelle vecchie definizioni deve essere >5000/mm3. Ormai quasi tutti gli apparecchi, i contaglobuli danno il numero assoluto dei linfociti e non soltanto la percentuale, comunque se non la danno basta che fate una semplice moltiplicazione e per fare una diagnosi di leucemia linfatica cronica occorrono più di 5000 linfociti per mm3 nel sangue periferico. Questo criterio è stato un pochettino rivisto negli ultimi anni, ma comunque sempre rimane abbastanza valido. Questi linfociti però devono essere caratterizzati da determinate caratteristiche, cioè non è che chiunque abbia più di 5000 linfociti ha una leucemia linfatica cronica, perché ad esempio anche un paziente con infezione da virus di Epstein-Barr potrebbe avere più di 5000 linfociti, anche uno che ha il CMV, quindi occorre definire bene questi linfociti per caratterizzarli come leucemia linfatica cronica. Questi linfociti sono caratterizzati dal fatto che se andiamo a guardare le immunoglobuline che stanno sulla superficie di questi linfociti sono immunoglobuline a bassa densità, cioè ci sono poche immunoglobuline rispetto a quelle che sono le immunoglobuline che noi troviamo nei linfociti normali. Questo perché sono un pochettino più immaturi nella ontogenesi linfocitaria rispetto ai linfociti normali, sono sempre linfociti abbastanza maturi, ma leggermente più immaturi dei linfociti normali, per cui la produzione di immunoglobuline è ancora non completa e queste immunoglobuline sono soprattutto IgM e IgD. Poi sono linfociti che vengono prodotti da un solo clone linfocitario perché è una patologia neoplastica quindi è una trasformazione neoplastica di un clone linfocitario e per definizione la trasformazione neoplastica deve essere monoclonale, è un solo clone che prolifera. Quindi mentre normalmente se andiamo a fare una valutazione delle catene leggere delle immunoglobuline nei linfociti normali troviamo sia linfociti che producono catene leggere λ che linfociti che producono catene leggere κ. Nel caso in cui noi andiamo ad esaminare i linfociti con leucemia linfatica cronica queste monteranno dentro il citoplasma soltanto catene di un solo tipo, o saranno tutte k o saranno tutte λ. Questo è uno dei cardini fondamentali di dimostrazione di monoclonalità che ci permette di differenziare appunto una linfocitosi reattiva da una linfocitosi neoplastica. Cioè se noi abbiamo tanti linfociti tutti uguali e sappiamo che sono tutti linfociti B, come facciamo a dire se quella linfocitosi è una linfocitosi reattiva o è una linfocitosi neoplastica? Il modo più semplice ed accurato è quello di andare a valutare le catene leggere κ e λ. Se noi vediamo che sono linfociti che hanno tutti quanti catene κ o tutti quanti catene λ abbiamo la conferma che si tratta di una popolazione linfoide monoclonale. Viceversa se ci sono ambedue le catene leggere equamente distribuite o più spesso sono distribuite in un rapporto 2:1 possiamo dire che quella è una linfocitosi non monoclonale. Nella leucemia linfatica cronica i linfociti appartengono alla linea B linfocitaria e quindi noi troveremo una positività per i marcatori per gli antigeni B linfocitari che sono soprattutto il CD19 e il CD20, anche il CD23 è positivo ma è meno importante. Una cosa importante è che questi linfociti di leucemia linfatica cronica presentano sulla loro superficie anche un altro antigene che si chiama CD5 che normalmente è espresso sui linfociti T per cui noi avremo una espressione aberrante di un marcatore T linfocitario in linfociti che peraltro sono linfociti B. Questo ci permette di fare la diagnosi differenziale anche con altri tipi di malattie linfoproliferative perché le uniche 2 malattie linfoproliferative che sono CD5-positive nell'ambito di una popolazione linfocitaria B sono la: − leucemia linfatica cronica; − linfoma mantellare. Quando noi troviamo un processo linfoproliferativo a carico dei B linfociti e quindi saranno CD19positivi e CD20-positivi e che monta pure il CD5 abbiamo sicuramente una diagnosi o di leucemia linfatica cronica o di linfoma mantellare. Poi ci sono altri aspetti che ci permettono di fare la diagnostica differenziale tra le due condizioni, ma già abbiamo una diagnosi ben precisa. Questa è una malattia che interessa ovviamente il midollo perché è lì che nascono i linfociti, che vengono prodotti e che poi passano nel sangue periferico, quindi nella definizione diagnostica della leucemia linfatica cronica occorre che ci sia una infiltrazione midollare più del 30%, cioè con una infiltrazione linfocitaria midollare superiore al 30%. (slide) Questi sono alcuni quadri morfologici della leucemia linfatica cronica. Vedete queste qua per esempio sono le cosiddette ombre di Gumprecht che sono abbastanza patognomoniche della leucemia linfatica cronica. Questi sono dei piccoli linfociti. Si tratta di uno striscio di sangue periferico di un paziente con leucemia linfatica cronica e vedete che ci sono linfociti maturi e in più ci sono queste cellule che non sono altro che delle cellule fragili che si rompono quando noi strisciamo il sangue sul vetrino e si chiamano ombre di Gumprecht. (slide) Questo è un grafico citofluorimetrico ed è caratterizzato dal fatto che su un asse ci sta il CD19 e sull'altro asse ci sta il CD5, quindi la positività del CD19 e del CD5 ci permette di identificare queste cellule come cellule appartenenti ad un paziente con leucemia linfatica cronica. Vi dicevo che c'è anche un interessamento midollare. (slide) Questa è una rappresentazione grafica del midollo osseo e delle trabecole ossee. Il colore più scuro rappresenta l'infiltrazione midollare di malattia che può essere una infiltrazione paratrabecolare, cioè può essere attorno alle trabecole ossee oppure può essere una infiltrazione nodulare o anche diffusa. La distribuzione della localizzazione midollare della leucemia linfatica cronica può essere abbastanza variabile. Usualmente la stragrande maggioranza dei linfomi, soprattutto i linfomi follicolari, hanno una localizzazione paratrabecolare di malattia, cioè vicino alle trabecole ossee. (slide) Rappresentazione di un nodulo midollare in un paziente con leucemia linfatica cronica. Quando noi vediamo una linfocitosi nel sangue periferico non è che tutte le linfocitosi appartengono alla leucemia linfatica cronica, anche se noi abbiamo la dimostrazione di monoclonalità possono esserci altre condizioni, altri linfomi per esempio che sono monoclonali e che prima danno una localizzazione osteo-midollare e successivamente da lì passano nel sangue periferico. Parliamo in questo caso di “linfoma leucemizzato”. Quindi il quadro clinico e morfologico di un linfoma leucemizzato può essere assolutamente sovrapponibile a quello di una leucemia linfatica cronica. Esistono anche altre condizioni per esempio che possono leucemizzarsi. (slide) Questa qui per esempio è la cosiddetta tricoleucemia. Questi linfociti presentano delle estroflessioni citoplasmatiche che li fanno assomigliare a dei capelli, per cui questa malattia linfoproliferativa si chiama tricoleucemia. (slide) Questa è invece la localizzazione leucemica di un linfoma follicolare, cioè un linfoma che prima ha invaso il midollo e poi è passato nel sangue periferico. Anche altre condizioni come per esempio un linfoma plasmocitoide, un linfoma splenico o anche una leucemia acuta possono dare tutti quadri molto simili sul piano morfologico alla leucemia linfatica cronica. Inoltre la leucemia linfatica cronica rappresentata da cellule molto mature si può anche trasformare in una sindrome di Richter cosiddetta che non è altro che la trasformazione di una leucemia linfatica cronica in un linfoma ad altro grado di malignità, usualmente in un linfoma a grandi cellule. Oppure si può trasformare in una leucemia cosiddetta prolinfocitica dove appunto questi linfociti che hanno un aspetto maturo assumono un aspetto molto più immaturo (si vede un grosso nucleolo) e prendono il nome di prolinfociti. Se ci sono più del 55% di queste cellule nel sangue periferico si chiama leucemia prolinfocitica, cioè una possibile evoluzione della leucemia linfatica cronica, anche se esistono le leucemia prolinfocitiche de novo. La leucemia linfatica cronica vi dicevo è una malattia dove noi troviamo un numero elevato di globuli bianchi, di linfociti nel sangue periferico, però questa malattia nasce nel midollo e nel midollo ha il suo nutrimento e in effetti queste cellule di leucemia linfatica cronica se noi le prendiamo e le mettiamo in coltura piano piano muoiono da sole. Se invece queste cellule le coltiviamo insieme a cellule dello stroma midollare, quindi a tutte le cellule che costituiscono il microambiente midollare vedete che queste cellule non muoiono. Questo perché appunto la fisiopatologia della leucemia linfatica cronica è caratterizzata dal fatto che queste cellule vengono protette nell'apoptosi e vengono supportate nella crescita dalle cellule stromali del midollo. È quindi nel midollo osseo che queste cellule crescono, si moltiplicano e poi da lì passano nel sangue periferico. Sono state identificate delle rare cellule che si possono anche isolare e poi espandere dal sangue periferico che vengono definite nurse-like cells, cioè delle cellule nutrici di queste cellule linfatiche croniche e che sono in realtà delle cellule macrofagiche che assumono questa funzione e mettendo in coltura le cellule di leucemia linfatica cronica senza queste nurse-like cells oppure con queste cellule, vedete che c'è una differenza nella vitalità di queste cellule di leucemia linfatica cronica che rimangono vitali se coltivate assieme a queste cellule nurse-like. Questo per darvi un attimo qualche informazione sulla fisiopatologia. Torniamo un po' alla clinica della leucemia linfatica cronica. La clinica della leucemia linfatica cronica è caratterizzata dal fatto che questi pazienti hanno una linfocitosi, quindi tutti i pazienti con leucemia linfatica cronica hanno la presenza di questi linfociti che sono B-linfociti CD5-positivi monoclonali con una restrizione monotipica κ o λ. Oltre alla linfocitosi possono avere anche un interessamento linfonodale, un interessamento epato-splenico oppure un deficit midollare per invasione midollare. Questo è molto importante perché indica una progressione della malattia che si associa anche a una differente prognosi, per cui i pazienti che hanno una linfocitosi nel sangue periferico con più di 5000 linfociti nel sangue periferico e un infiltrazione midollare più del 30% queste configurano la diagnosi di leucemia linfatica cronica a patto che noi dimostriamo che sono monoclonali, quindi che hanno una restrizione κ o λ, che sono linfociti B con la presenza del CD5 e quindi abbiamo la tipica cellula di leucemia linfatica cronica con la linfocitosi e l'invasione midollare. Questi pazienti non hanno nessun'altra localizzazione di malattia, quindi visitando questi pazienti noi non troviamo nulla, non troviamo adenopatie, non troviamo epato-splenomegalie, nulla. Sono pazienti che non sono anemici e non sono piastrinopenici. − Questi pazienti si definiscono stadio zero (0) secondo questa classificazione di RAI (RAI è un indiano che lavora negli Stati Uniti e che ha fatto questa classificazione). − Se accanto alla linfocitosi troviamo anche che questi pazienti hanno delle tumefazioni linfonodali, linfoadenopatie, ovunque ce l'abbiano, questo identifica il paziente in stadio I di RAI. − Se oltre alla linfocitosi è presente o splenomegalia o epatomegalia si definisce stadio II. Ci può essere in questo caso anche la linfoadenopatia, ma non è necessario che ci sia la linfoadenopatia per fare diagnosi di stadio II. − Se c'è la linfocitosi più l'anemia e l'emoglobina inferiore a 11, con o senza le precedenti localizzazioni che abbiamo visto si definisce stadio III. − Se c'è linfocitosi più piastrinopenia, meno di 100 000 piastrine con o senza tutto quello che abbiamo visto prima, stadio IV. Quindi il denominatore comune è la linfocitosi, alla quale si aggiunge poi o i linfonodi ingrossati o fegato e milza ingrossati o anemia e piastrinopenia che sono la conseguenza di una infiltrazione midollare massiva che impedisce la produzione di globuli rossi e di piastrine. Questa classificazione molto semplice e molto grossolana ci permette però anche di fare una valutazione prognostica perché vedete come la sopravvivenza mediana dei pazienti in stadio 0 è più di 10 anni, mentre la sopravvivenza mediana dei pazienti che sono anemici o piastrinopenici e di 1,5 anni. Gli stadi avanzati di malattia hanno una sopravvivenza paragonabile a quella di una leucemia acuta quasi, mentre gli stadi iniziali di malattia hanno una sopravvivenza che, considerando che si tratta di pazienti anziani, con una sopravvivenza di più di 10 anni, è una sopravvivenza molto ragguardevole. Quindi parliamo di pazienti che hanno la stessa malattia, che hanno tutti la leucemia linfatica cronica ma con una prospettiva prognostica totalmente differente. Lo stesso vale per quest'altra classificazione che è un pochettino più semplificata che è la classificazione di Binet ( un francese che ha fatto questa classificazione) che è basata sugli stessi concetti detti in precedenza, ma è più semplificata, forse anche più facile da ricordare. Classificazione di Binet: Stadio C: pazienti in stadio più avanzato, anemici o piastrinopenici. L'unica differenza è che il cut-off di emoglobina nella precedente classificazione era 11g qui è 10g, ma fondamentalmente il concetto è lo stesso, ossia che il paziente anemico e piastrinopenico è un paziente con malattia più avanzata e che quindi ha prognosi sfavorevole. Gli stadi iniziali sono i pazienti che non hanno anemia e non hanno piastrinopenia e hanno meno di 3 aree coinvolte, dove le aree vengono considerate le stazioni linfonodali latero-cervicali, ascellari e inguinali più fegato e milza, quindi al massimo uno può avere 5 aree interessate. Se uno ha meno di tre, quindi o una o due di queste aree interessate allora viene definito come stadio A, senza che ha anemia e piastrinopenia. Se invece ha tre o più di queste aree interessate viene definito stadio B. Lo stadio A ha una sopravvivenza mediana che è uguale a quella dei pazienti della stessa età e sesso che non hanno malattia! Quindi un paziente con leucemia linfatica cronica in stadio A è come se la malattia non l'avesse perché ha una sopravvivenza assolutamente uguale a quella di una popolazione di pari età e sesso che non ha malattia. Poi bisogna fare dei distinguo che vedremo successivamente, però in linea di massima avere una leucemia linfatica cronica in stadio A significa non avere una malattia e la grande maggioranza di questi pazienti non vanno trattati perché non hanno una malattia, hanno solo una condizione che potrebbe diventare una malattia nel caso ci sia una evoluzione verso gli stadi più avanzati. Vedo molti pazienti con leucemia linfatica cronica in stadio A e dico di non fare nessuna terapia e sia il paziente che il medico curante rimangono un pochettino perplessi, ma questa condizione di leucemia ha solo il nome, perché non è una vera e propria leucemia, è una condizione neoplastica che non progredisce, che non determina nessuna compromissione dell'organismo e soprattutto non determina nessuna riduzione dell'aspettativa di vita, tranne per un sottotipo che voi vedremo successivamente. Viceversa la sopravvivenza di pazienti con anemia o piastrinopenia è una sopravvivenza molto breve, quindi sicuramente questi pazienti vanno trattati e vanno trattati anche in maniera abbastanza aggressiva. Un discorso intermedio va fatto per i pazienti che hanno una mediana di sopravvivenza di 7 anni e quindi tutto dipende dall'età del paziente, dalle condizioni ecc ecc Vi dicevo appunto che la maggior parte dei pazienti in stadio A sono pazienti che non vanno in progressione, però c'è una piccola quota di questi pazienti che va in progressione e allora si è sempre cercato di identificare questi pazienti che sono in stadio A semplicemente perché noi abbiamo fatto una diagnosi molto precocemente ma in realtà hanno una malattia che tende alla progressione. Così ci sono delle vecchie definizioni, per esempio i pazienti che hanno meno di 30 000 globuli bianchi e più di 12g di emoglobina vengono definiti come stadio A1, dove la probabilità di progressione è veramente bassissima. Mentre i pazienti che hanno un emoglobina un po' più bassa, hanno un po' più linfociti, hanno una infiltrazione midollare piuttosto marcata, che hanno un numero di aree interessate inferiori a 2, tutti questi sono pazienti che hanno una maggiore probabilità di progressione e quindi vanno seguiti con un po' più attenzione per cogliere eventuali progressioni. Per cui lo stadio A “smoldering” ha una bassissima probabilità di andare incontro a una progressione, contro uno stadio A un pochettino più attivo che ha una maggiore probabilità di andare incontro a una progressione. Ma adesso abbiamo delle modalità differenti per valutare quelle che sono le possibilità di progressione di questi pazienti perché conosciamo meglio la biologia di questa malattia e quindi non ci basiamo soltanto sull'esame clinico come appunto le vecchie classificazioni di RAI o di Binet ci impongono, queste sono chiaramente la prima tappa nella valutazione del paziente con leucemia linfatica cronica e sono le cose più semplici, più grossolane, basta visitare il paziente e già lo abbiamo definito secondo la classificazione di RAI o di Binet. Quello che adesso abbiamo imparato è che ci sono delle alterazioni citogenetiche che possono configurare una ben specifica prognosi nei pazienti con leucemia linfatica cronica. Appunto facendo la citogenetica a questi pazienti si è visto come i pazienti che hanno una delezione del 17 vanno molto male e vanno male anche i pazienti che hanno la delezione dell'11, mentre i pazienti che hanno la trisomia del 12 o che hanno la delezione del 13 vanno o come i pazienti che non hanno alcuna anomalia citogenetica oppure addirittura meglio, quelli che hanno la delezione del 13 vanno meglio di quelli con cariotipo normale. (slide) Quindi possiamo definire da questa curva che le alterazioni citogenetiche sfavorevoli in questi pazienti con leucemia linfatica cronica sono la delezione dell'11 e la delezione del 17. Per l'11 non sappiamo bene per quale motivo questi pazienti vanno male, il 17 è abbastanza intuibile perché nel 17 ci sta la P53. I pazienti in cui manca la P53 sono pazienti che accumulano molte alterazioni molecolari, molte alterazioni del loro genoma e quindi queste cellule diventano particolarmente maligne. Quindi noi sicuramente al di là dello stadio di malattia possiamo ben dire che i pazienti con leucemia linfatica cronica possono avere un decorso poco aggressivo o molto aggressivo a seconda del tipo di alterazione citogenetica che presentano, per cui i pazienti che hanno la delezione del 13 vanno molto bene, i pazienti che hanno la delezione del 17 vanno molto male, quelli che hanno la delezione dell'11 vanno male, quelli che hanno la trisomia del 12 vanno grossomodo come pazienti che hanno un cariotipo normale, quindi molto importante in questo caso il cariotipo dei pazienti con leucemia linfatica cronica perché ci fa predire la prognosi di questi pazienti. Per cui se noi troviamo per esempio un paziente che è in stadio A di malattia, ma che presenta una delezione del 17 corriamo subito ai ripari, anche se è una cosa abbastanza rara perché la delezione del 17 ce l'hanno i pazienti che hanno una malattia più avanzata. Qual è il decorso clinico di questi pazienti con leucemia linfatica cronica? Questi pazienti usualmente hanno una malattia estremamente indolente e possono non avere nessun sintomo, soprattutto i pazienti in stadio A non hanno alcun sintomo. Gli altri pazienti possono avere degli altri sintomi, in particolare i sintomi che sono legati all'ingrossamento dei linfonodi o all'anemia. Quando la malattia va in progressione ci può essere anche un peggioramento della funzione midollare, quindi peggioramento dell'anemia e peggioramento della piastrinopenia e si può avere anche un ingrossamento particolare della milza con condizione di ipersplenismo. E poi possono accompagnarsi quegli altri sintomi che sono tipici delle malattie linfoproliferative, soprattutto appunto la febbre e la perdita di peso. Una cosa importante è che in questi pazienti c'è una elevata frequenza di malattie autoimmunitarie e questo non dipende dal fatto che questi linfociti producono immunoglobuline ma dipende dal fatto che questa condizione, come ho detto l'altro volta per i linfomi a basso grado di malignità, determina uno sbilanciamento del sistema immunitario per cui vengono meno i linfociti T regolatori che permettono lo sviluppo di cloni autoreattivi e quindi questi cloni autoreattivi, che sono cloni normali, non sono cloni neoplastici, sono linfociti normali, producono anticorpi contro i propri costituenti e non sappiamo bene per quale motivo ma uno dei bersagli più frequenti sono i globuli rossi. È molto frequente avere una anemia emolitica autoimmune. Questo in qualche modo complica la situazione perché avete visto come una delle condizioni che fa passare di stadio la leucemia linfatica cronica è proprio l'anemia. L'anemia identifica il terzo stadio di RAI e lo stadio C di Binet, cioè lo stadio più avanzato. Questo perché questa anemia è legata all'infiltrazione midollare, quanta più infiltrazione midollare c'è, quanto più il midollo non produce globuli rossi e quindi diventa anemico il paziente. Ma avendo questi il paziente pure la possibilità di malattie autoimmuni e quindi di anemia emolitica autoimmune, un'altra possibile condizione che determina l'anemia in questi pazienti è appunto l'anemia emolitica autoimmune. Quindi ci sono almeno 2 condizioni che possono provocare l'anemia in questi pazienti: − infiltrazione midollare; − anemia emolitica autoimmune. L'anemia emolitica autoimmune però non configura uno stadio avanzato di malattia, configura semplicemente una complicazione della leucemia linfatica cronica e in questo caso è abbastanza semplice fare la diagnosi differenziale tra le due condizioni, cioè una condizione in cui c'è una infiltrazione midollare che determina la mancata produzione di globuli rossi e l'altra condizione in cui c'è una distruzione eccessiva di globuli rossi. Qual è l'esame che bisogna fare per fare questa diagnosi differenziale? I reticolociti! Fare la conta dei reticolociti ci permette di fare questa diagnosi differenziale. È chiaro poi che i pazienti che hanno una anemia emolitica autoimmune avranno tutti gli altri segni dell'emolisi, avranno la bilirubina indiretta aumentata, avranno LDH aumentato, l'aptoglobina bassa ecc.. ma l'esame più importante nella diagnostica differenziale è la conta dei reticolociti. Perché chiaramente se un'anemia è dovuta ad una infiltrazione midollare avremo una bassa conta reticolocitaria, viceversa se l'anemia è dovuta a una condizione di emolisi periferica avremo un'alta conta reticolocitaria. Quindi questo è un esame che ci permette subito di fare una diagnosi differenziale però chiaramente capite come il paziente può essere anemico per diversi motivi. Un'altra condizione importante che si associa spesso alla leucemia linfatica cronica e che è espressione appunto di questo disordine immunologico generalizzato è il fatto che c'è una ipogammaglobulinemia e quindi questi pazienti sono proni a sviluppare delle infezioni. Quindi come vi dicevo non tutti i pazienti affetti da leucemia linfatica cronica vanno sottoposti al trattamento, anzi la stragrande maggioranza di questi pazienti non deve essere trattata perché hanno una malattia limitata in stadio 0 di RAI o stadio A di Binet. Però ci sono delle condizioni in cui è necessario iniziare il trattamento e chiaramente questo trattamento va iniziato quando: − ci sono sintomi B, cioè sintomi legati alla malattia linfoproliferativa come la febbre, la sudorazione notturna, la perdita di peso; − c'è una linfocitosi molto elevata che si accompagna soprattutto ad un deficit di produzione da parte del midollo, quindi anemia e piastrinopenia; − presenza di una anemia emolitica autoimmune o anche di una trombocitopenia autoimmune, perché gli anticorpi nella stragrande maggioranza dei casi sono diretti contro i globuli rossi, ma talvolta anche contro le piastrine; − presenza di massiva splenomegalia; − malattia “bulky”, nel senso di linfonodi molto grossi; − la compromissione del sistema immunitario è tale per cui il paziente ha delle ricorrenti infezioni batteriche. Vi dicevo comunque come questi pazienti siano frequentemente bersaglio di una condizione autoimmune e l'anemia emolitica autoimmune interviene in circa il 15% dei casi e addirittura la positività al test di Coombs che è il test che ci permette di evidenziare gli autoanticorpi contro i globuli rossi. Esiste il test di Coombs diretto ed indiretto: il diretto è quello che ci fa vedere gli anticorpi adesi ai globuli rossi, l'indiretto è quello invece che ci fa vedere i globuli rossi circolanti, presenti nel siero. Questo test di Coombs è positivo nel 30% dei pazienti, quindi molti pazienti hanno gli autoanticorpi contro i globuli rossi anche se poi non tutti i pazienti sviluppano clinicamente l'anemia emolitica autoimmune. Ci può essere una trombocitopenia o più raramente una granulocitopenia sempre legata a degli anticorpi e questo dipende non tanto dai linfociti neoplastici quanto dalla presenza di questi cloni autoreattivi. Abbiamo visto che è molto importante valutare alcuni aspetti biologici dei pazienti con leucemia linfatica cronica per predirne la prognosi. In particolare è molto importante valutare la citogenetica, ma non solo la citogenetica, ci sono anche altri fattori. (slide) Questo è ad esempio uno studio che il prof ha fatto sulla timidina chinasi. Vedete i pazienti che avevano una timidina chinasi bassa andavano meglio rispetto a quelli che avevano una timidina chinasi alta, ma questo è abbastanza logico perché la timidina chinasi è un marcatore di proliferazione, quindi i pazienti che hanno una malattia che prolifera più rapidamente chiaramente vanno peggio rispetto a chi ha una malattia che prolifera più lentamente. Di recente abbiamo però scoperto un altro aspetto, che in realtà sul piano biologico questa leucemia linfatica cronica si può dividere in 2 condizioni importanti: − una malattia che origina da cellule che hanno incontrato l'antigene nel centro germinativo; − una malattia che deriva da cellule, sempre linfociti B, che non hanno incontrato l'antigene. E questo si vede attraverso l'analisi mutazionale delle immunoglobuline. Se facciamo una analisi mutazionale delle immunoglobuline e vediamo che c'è stato un riarrangiamento VDJ, vuol dire che quelle cellule hanno incontrato l'antigene, se invece il VDJ non è riarrangiato vuol dire che quelle cellule non hanno incontrato l'antigene. E questo è molto importante perché è stato visto che i pazienti le cui cellule di leucemia linfatica cronica sono cellule che hanno incontrato l'antigene, che quindi sono passate dal centro germinativo e hanno incontrato l'antigene e poi c'è stata questa trasformazione in leucemia linfatica cronica, queste vanno molto meglio rispetto a quelle leucemie linfatiche croniche che derivano da cellule che non hanno incontrato l'antigene e che quindi hanno un pattern di immunoglobuline non mutato. Quindi è molto importante adesso fare questa valutazione, se queste cellule di leucemia linfatica cronica hanno un pattern di riarrangiamento dei geni delle immunoglobuline di tipo mutato o non mutato. Quelli che originano da cellule che hanno un pattern non mutato vanno peggio rispetto a quelli che hanno un pattern mutato. Accanto a questo, siccome la valutazione del riarrangiamento genico delle immunoglobuline è abbastanza complessa, si è cercato anche di identificare dei marcatori biologici di più semplice valutazione che potessero fare da surrogato di queste informazioni che ci vengono da questi studi biologici particolarmente sofisticati. Uno di questi marcatori si è dimostrato il CD38, cioè andando a valutare con la citofluorimetria su queste cellule la positività o la negatività del CD38, questo è un altro marcatore prognostico importante che correla molto con lo stato delle immunoglobuline mutato o non mutato, cioè i pazienti che sono CD38-positivi vanno peggio rispetto ai CD38-negativi perché c'è una correlazione tra l'espressione del CD38 e lo stato delle immunoglobuline mutato o non mutato. I pazienti non mutati che abbiamo visto sono quelli che vanno peggio, hanno pure l'espressione del CD38, anche se purtroppo non c'è una concordanza completa. (slide) Vedete qui è rappresentato il CD38 e qui lo stato mutato o non mutato e vedete che molti pazienti che hanno un pattern immunoglobulinico non mutato hanno una positività per il CD38 e viceversa molti pazienti con lo stato immunoglobulinico mutato hanno una negatività per CD38, ma esiste una quota non trascurabile di pazienti che vanno esattamente al contrario, cioè hanno una positività per il CD38 pure essendo mutati e una negatività per il CD38 pur essendo non mutati. Quindi esiste una correlazione piuttosto stretta tra stato mutazionale delle immunoglobuline ed espressione del CD38, ma non è una correlazione definitiva. E i pazienti che hanno uno stato mutazionale di tipo mutato e sono CD38-negativi vanno molto meglio rispetto a chi ha uno stato mutazionale di tipo non mutato ed è CD38-positivo. Poi “nel mezzo” ci sono quelli che sono intermedi, cioè quelli che sono discordanti che possono avere una positività di CD38 e stato mutazionale di tipo mutato o viceversa non mutati e CD38negativi. È una vicenda un po' complessa, ma ricordate che la configurazione delle immunoglobuline di tipo non mutato configura una prognosi peggiore rispetto al mutato e la positività per il CD38 è una prognosi peggiore rispetto alla negatività per il CD38. Un altro parametro che dovrebbe essere di semplice valutazione è lo Zap70. Anche qui lo Zap70 correla con lo stato non mutato. (slide) Qui vedete è una valutazione di “gene expression profiling” in pazienti con una condizione delle immunoglobuline di tipo non mutato versus i pazienti di tipo mutato e vedete che molti di questi pazienti hanno una positività per lo Zap70 che è una proteina che si può identificare anche in citofluorimetria, per cui questo ha grossomodo lo stesso valore del CD38. I pazienti con una configurazione immunoglobulinica di tipo non mutato sono pure Zap70positivi e i pazienti invece con una configurazione immunoglobulinica di tipo mutato sono invece Zap70-negativi. Quindi questi due parametri, Zap70 e CD38, vengono utilizzati spesso al posto delle immunoglobuline. Noi adesso cerchiamo di fare un pochettino tutti questi parametri, cioè facciamo lo stato mutazionale, mutato o non mutato, facciamo lo Zap70, facciamo il CD38 per cercare di avere quante maggiori informazioni possibili su quella che è la prognosi di questi pazienti perché tutti questi parametri sono spesso correlati tra di loro, ma hanno una certa indipendenza. (slide) Anche qui vedete pazienti che sono Zap70-positivi e hanno un pattern immunoglobulinico non mutato vanno molto peggio rispetto a pazienti che sono Zap70negativi e sono mutati. Quindi noi quando vediamo un paziente con leucemia linfatica cronica non possiamo più limitarci a visitare il paziente e fare una valutazione secondo la stadiazione di RAI o di Binet, se è un paziente in stadio iniziale o in stadio avanzato, perché quello è vero, è sacrosanto ma è molto grossolano. C'è bisogno adesso di inquadrare meglio il paziente e di capire quella che è la sua prognosi. (slide) Abbiamo pazienti tutti con leucemia linfatica cronica. I pazienti che hanno un pattern di riarrangiamento dei geni delle immunoglobuline di tipo mutato (più del 2% del riarrangiamento vuol dire che il linfocita ha incontrato l'antigene e c'è stato il riarrangiamento dei geni delle immunoglobuline, del VDJ, per cui si può considerare mutato) e che hanno un cariotipo normale oppure che hanno soltanto una delezione del l3. I pazienti con queste caratteristiche biologiche devono essere pazienti che hanno uno stadio A di Binet o 0 di RAI, che hanno un tempo di raddoppiamento dei linfociti che è superiore a 12 mesi, cioè noi facciamo l'emocromo oggi e il paziente ha 5000 linfociti, prima che arriva a 10 000 linfociti senza nessuna terapia passano più di 12 mesi. Questo è un altro parametro prognostico importante. Questi sono pazienti che hanno una b2-microglobulina normale, che hanno lo Zap70 negativo, che hanno il CD38 negativo. Questi sono pazienti che hanno un range di sopravvivenza prevista tra i 15 e i 20 anni. Se si tratta di pazienti settantenni capite bene che la loro prospettiva di vita è una prospettiva assolutamente normale, quindi questi pazienti non vanno assolutamente trattati, ma devono avere tutte queste caratteristiche, cioè dobbiamo studiare molto bene il paziente per rassicurarlo e rassicurarci che non ci sarà nessuna progressione di malattia. Quindi questi pazienti non possono essere seguiti in un posto qualunque, devono essere seguiti in ematologia dove noi siamo in grado di fare tutte queste valutazioni perché ricordate che questi sono pazienti che stanno bene, non hanno nessuna sintomatologia. Poi ci sono pazienti che hanno una prognosi intermedia e sono pazienti che possono avere pattern dei geni delle immunoglobuline di tipo non mutato ( meno del 2% di riarrangiamento), sono pazienti che possono avere la delezione dell'11 o del 6 o una trisomia del 12. Quindi sono i pazienti che hanno uno stadio di Binet o di RAI leggermente più avanzato, stadio B di Binet o I-II di RAI, possono avere un tempo di raddoppiamento dei linfociti inferiore a 12 mesi, possono avere una alta b2-microglobulina, possono avere uno di questi fattori positivi, Zap70 e CD38. Questi sono sicuramente pazienti che vanno trattati perché hanno una sopravvivenza prevista tra i 5 e i 10 anni. È chiaro poi che sei un paziente ha 85 anni con queste caratteristiche non lo trattiamo. Poi ci sono i pazienti peggiori. I pazienti peggiori sono tutti i pazienti che hanno la delezione del 17, ricordate che questa è una condizione estremamente negativa, ma per fortuna anche estremamente rara, non più del 5% dei pazienti con leucemia linfatica cronica presenta questa delezione del 17, però quelli che ce l'hanno vanno malissimo. Sono pazienti che hanno sempre uno stato non mutato, possono avere anche la delezione dell'11, sono pazienti in stadio di malattia avanzato, quindi stadio C di Binet e stadio II-IV di RAI. Sono pazienti che hanno un tempo di raddoppiamento molto rapido, hanno una b2microglobulina alta. Hanno una positività per Zap70 e CD38. È chiaro che queste sono delle classificazioni un po' rigide. I pazienti possono avere delle caratteristiche che sono trasversali e quindi chiaramente bisogna poi valutare il peso di ogni singola alterazione. Per esempio il peso del riarrangiamento dei geni delle immunoglobuline è molti più importante di quello che può essere il peso ad esempio del CD38 in termini di prognosi. Oppure la citogenetica ancora di più: uno che ha la delezione del 17, qualunque altra caratteristica clinico-laboratoristica abbia, quella che comanda è la delezione del 17. Domanda di una collega: “Ma se non hanno nessun sintomo perché vengono da noi?” Risposta: “Perchè fanno un esame di laboratorio, fanno un emocromo e vedono che c'è una linfocitosi, questo è usualmente il motivo. Si tratta di pazienti anziani e il paziente anziano magari ha un altro motivo per farsi un esame, per esempio ha una ipertrofia prostatica, il medico gli richiede il PSA e gli richiede nel frattempo pure un emocromo e quindi si scopre che ha questa linfocitosi. La maggior parte dei pazienti con leucemia linfatica cronica vengono dall'ematologo per un esame laboratoristico fatto occasionalmente, per una linfocitosi scoperta occasionalmente.” Vediamo qualche cenno di terapia. I farmaci che noi utilizziamo al momento per la leucemia linfatica cronica sono la Fludarabina, che è un analogo delle purine e il Chlorambucil che è un alchilante. La Fludarabina è un farmaco molto più forte, molto più aggressivo e con maggiori effetti collaterali, per cui generalmente i pazienti giovani li trattiamo con Fludarabina in combinazioni con altri farmaci, mentre i pazienti anziani li trattiamo con il Chlorambucil. Anche il Rituximab che vi ho fatto vedere l'altra volta è un farmaco che viene utilizzato molto nel trattamento dei linfomi non-Hodgkin e viene anche utilizzato nel trattamento dei pazienti con leucemia linfatica cronica e in questo caso l'effetto è sinergico per cui la terapia più utilizzata al momento è la combinazione di Fludarabina, Ciclofosfamide e Rituximab che è quella che al momento ci dà la maggiore percentuale di risposte in questi pazienti affetti da leucemia linfatica cronica. (slide) Se vedete questa curva di sopravvivenza nei pazienti trattati con Fludarabina da sola, Fludarabina più Ciclofosfamide o Mitoxantone (quindi Fludarabina più un altro chemioterapico), se aggiungiamo il Rituximab otteniamo una curva di sopravvivenza che indica un maggior guadagno. Questa curva non è ancora lunga come le altre curve perché ancora la possibilità di utilizzare il Rituximab in questa patologia ce l'abbiamo solo da qualche anno. Tuttavia la Fludarabina, come tutti gli analoghi delle purine, ha un grosso svantaggio e lo svantaggio è quello di determinare una forte depressione dei linfociti T, è molto tossica per i linfociti T. Oltre ad essere efficace nei confronti dei linfociti B neoplastici, purtroppo distrugge anche i linfociti T normali e i pazienti che fanno Fludarabina hanno una forte depressione sia dei CD4 che dei CD8. E per parecchi mesi, 24 mesi (slide), ancora c'è una forte depressione dei CD4 e dei CD8. Tutto questo comporta un rischio importante in termini di infezione. Il prof ammette che il prof Russo gli manda sempre maledizioni perché i pazienti trattati con Fludarabina poi finiscono nei reparti di malattie infettive. La Fludarabina quindi e tutti gli analoghi delle purine determinano una forte immunodepressione. Questa molecola determina anche una risposta terapeutica importante per questi pazienti che hanno un prolungamento significativo della sopravvivenza però quando poi il paziente arriva al capolinea questa condizione di immunodepressione si fa sentire per cui sono pazienti che sviluppano numerose infezioni opportunistiche. Oltre a questo, questo sbilanciamento del sistema immunitario favorisce anche lo sviluppo di anemie emolitiche autoimmuni. Vi ho già detto che la anemia emolitica autoimmune fa parte delle possibili complicazioni dei pazienti affetti da leucemia linfatica cronica. I pazienti trattati con Fludarabina hanno un maggiore rischio di sviluppare anemie emolitiche autoimmuni, proprio per uno sbilanciamento della normale popolazione linfocitaria. Quindi se da un lato questi farmaci sono molto attivi nei confronti della malattia leucemica, dall'altro provocano tutta una serie di effetti collaterali importanti. Un altro farmaco che viene utilizzato nella leucemia linfatica cronica in pazienti di nicchia, fondamentalmente lo utilizziamo soltanto nei pazienti che hanno la delezione del 17, i più gravi, i più a rischio, è l'anti-CD52, un anticorpo rivolto verso un antigene espresso praticamente su tutti i linfociti. Questo farmaco è in grado di determinare una forte riduzione delle cellule leucemiche. (slide) Vedete qui una biopsia osteo-midollare di un paziente affetto da leucemia linfatica cronica prima e dopo il trattamento come è netta la differenza. Però vedete come l'Alemtuzumab (anti-CD52) determina una spiccata riduzione dei CD4 e questa deplezione di CD4 si mantiene per un lungo periodo di tempo e questo vale anche per il CD8. Questi pazienti assumono un assetto linfocitario che è come quello dei pazienti con HIV, anzi peggiore, perché c'è una forte deplezione sia dei CD4 che dei CD8. Tutto questo espone ad un rischio infettivo, oltre che ad un rischio legato alle infusioni e alla mielosoppressione. Noi facciamo la profilassi con il Bactrim per evitare le infezioni batteriche, per evitare le infezioni da Pneumocystis. La profilassi che facciamo noi è un po' peculiare. Quello che ci spaventa di più quando facciamo la terapia con l'anti-CD52, l'Alemtuzumab, sono le riattivazioni da CMV, per cui costantemente una volta alla settimana facciamo il monitoraggio della viremia per CMV e contemporaneamente facciamo una profilassi con degli antivirali, in particolare noi utilizziamo il Ganciclovir proprio per evitare la progressione della viremia da CMV che è la condizione più frequente che si viene a verificare. La profilassi con il Bactrim fino a sei mesi dopo il trattamento con questi farmaci. La profilassi per il CMV la continuiamo fino ad un mese dopo che abbiamo finito la terapia con l'Alemtuzumab. Nonostante questo molti pazienti trattati con Alemtuzumab e con anti-CD20 si fanno le infezioni. Queste infezioni sono molto spesso legate ad una antigenemia da CMV, ma vedete che ci sono molte polmoniti ma anche molte febbri di origine sconosciuta. Infine un'altra possibilità terapeutica con leucemia linfatica cronica è il trapianto allogenico, non l'autotrapianto perché vedete qui (slide) l'autotrapianto non è caratterizzato da un plateaux, cioè no guarisce nessuno con l'autotrapianto, mentre guariscono i pazienti con leucemia linfatica cronica trattati con allotrapianto. Noi non abbiamo ancora parlato del trapianto, ma è una procedura limitata ad una ristrettissima nicchia di pazienti: devono essere pazienti particolarmente giovani, ma la leucemia linfatica cronica è una malattia dell'anziano. L'età media del paziente con leucemia linfatica cronica è 65-70 anni. Il trapianto allogenico è confinato a pazienti di età non superiore a 45-50 anni, quindi capite bene che la quota di pazienti che può essere trapiantata è molto piccola, poi ci sono degli altri problemi di compatibilità ecc Però negli anni c'è un progressivo guadagno in termini di sopravvivenza nella leucemia linfatica cronica. Questa è una diapositiva che viene dal M.D. Anderson Hospital di Houston, che è uno dei templi del trattamento della leucemia linfatica cronica dove appunto si vede come negli anni passati, prima dell'85, poi prima dell'89, poi prima del ‘99 e infine prima del 2003 c'è stato un progressivo guadagno mano a mano che sono entrati a nostra disposizione farmaci come la Fludarabina e anticorpi monoclonali come il Rituximab, il Campath. Tutto questo ha permesso un progressivo guadagno in termini di sopravvivenza per questa malattia che però rimane ancora una malattia dove non esiste un plateaux, non esiste una guarigione se non in quei rarissimi casi di pazienti che vengono sottoposti a trapianto di midollo allogenico. Lezione 10 MIELOMA MULTIPLO Il mieloma multiplo è una patologia neoplastica che interessa le plasmacellule, quindi c'è una proliferazione monoclonale di plasmacellule che usualmente avviene a livello del midollo osseo e queste plasmacellule, che in condizioni normali producono immunoglobuline, quando sono neoplastiche e quindi sono monoclonali producono tutte quante un uguale tipo di immunoglobulina. Ecco che noi vediamo al protidogramma elettroforetico la presenza di una componente anomala monoclonale di proteine che sono tutte uguali e quindi migrano in un punto elettroforetico e danno un picco al protidogramma a banda stretta perché sono tutte confinate in quel punto isoelettrico dell'elettroforesi. Questa malattia è inoltre caratterizzata da un'importante malattia ossea: (slide) vedete qui un cranio tutto “bucherellato” per la presenza di manifestazioni osteolitiche e qui c'è appunto una compromissione osteo-midollare con una iperattivazione osteoclastica. È caratterizzato inoltre da una frequente compromissione renale e come la maggior parte delle malattie linfoproliferative, essendoci una compromissione del sistema immunitario, questi pazienti sono esposti ad infezioni. (slide) Qui vedete in particolare una radiografia del torace che mostra un processo broncopneumonico. Quindi queste sono le caratteristiche più importanti del mieloma multiplo, una proliferazione monoclonale di plasmacellule che determina quasi sempre: - una alterazione al protidogramma, cioè all'elettroforesi, delle proteine del siero; - che determina spesso una malattia ossea; - che determina spesso una malattia renale; - che determina spesso un deficit immunologico tale che i pazienti sviluppano delle infezioni. Non abbiamo molte informazioni sulla causa dello sviluppo di mieloma multiplo, però certamente le radiazioni sono state implicate nello sviluppo del mieloma multiplo, tanto è che per esempio i vecchi radiologi, i vecchi tecnici di radiologia hanno una maggiore incidenza di sviluppare il mieloma multiplo, ma sicuramente una causa che adesso viene sempre più accreditata è una esposizione eccessiva agli erbicidi e ai pesticidi che si usano in agricoltura, per cui il mieloma multiplo è molto frequente nelle zone rurali soprattutto dove si fa un uso inadeguato dei diserbanti, degli erbicidi e dei pesticidi. Molto spesso appunto i nostri contadini non usano le adeguate protezioni e vengono esposti in modo eccessivo a queste sostanze che sono sicuramente in grado di favorire lo sviluppo del mieloma multiplo. Vi dicevo che il mieloma multiplo non è altro che la trasformazione neoplastica di elementi plasmacellulari. In realtà sul piano morfologico sono abbastanza limitate le differenze che esistono tra una plasmacellula di mieloma multiplo e una plasmacellula normale. Ci sono però delle differenze di ordine antigenico, in particolare se sia le plasmacellule normali e le plasmacellule neoplastiche sono CD38-positive, le plasmacellule normali sono più spesso CD56negative e CD19-positive, al contrario le plasmacellule mielomatose sono CD56-positive e CD19-negative. Queste sono delle caratteristiche antigeniche importanti, perché poi in realtà è molto difficile differenziare una plasmacellula normale da una plasmacellula di mieloma su un piano morfologico. Ci si riesce su un piano di citofluorimetria, ma è abbastanza difficile perché queste caratteristiche di positività e negatività sono abbastanza generali e non sono del tutto mantenute in tutte le plasmacellule. È molto importante considerare il fatto che la patogenesi della malattia del mieloma multiplo è un po' come un tavolino a tre piedi, dove un primo piede è rappresentato dalle cellule mielomatose, dalla neoplasia stessa, un altro piede è rappresento dalle cellule di supporto, dalle cellule stromali, dal microambiente midollare e di supporto appunto alle cellule mielomatose e un altro punto importante è il sistema degli osteoclasti e degli osteoblasti. Per cui è proprio l'interazione che esiste tra questi tre sistemi che determina lo sviluppo patogenetico e clinico delle manifestazioni cliniche del mieloma. Sicuramente queste plasmacellule crescono in maniera incontrollata rispetto alle plasmacellule normali e uno dei fattori più importanti che serve a far crescere queste plasmacellule è la interleuchina 6. È stato dimostrato un ruolo fondamentale dell'IL-6 nel mieloma multiplo: - innanzitutto l 'IL-6 inibisce l'apoptosi sia spontanea che indotta da FAS per esempio di queste plasmacellule e se incubiamo queste cellule con anticorpi anti-IL-6 vediamo che c'è una inibizione della proliferazione delle cellule mielomatose. Lo stesso se utilizziamo degli antisenso anti-IL-6. In laboratorio dei topi knockout per IL-6 non sviluppano mieloma multiplo che può essere indotto invece nei topi che hanno il gene per IL-6. - i livelli di IL-6 e del suo recettore correlano con l'attività di malattia - infine terapia con anticorpi anti-IL-6 riducono anche se transitoriamente le manifestazioni della malattia. Quindi tutte queste informazioni ci dicono che l'IL-6 è un fattore cruciale, un fattore molto importante nella proliferazione delle plasmacellule mielomatose e quindi nella crescita del mieloma e sono stati dimostrati due modelli, un modello autocrino e un modello paracrino. In entrambi i casi è l'IL-6 che funge da motore che fa proliferare queste cellule perché determina una attivazione appunto dei fattori di trascrizione ecc e nel caso più frequente, il modello paracrino, è l'IL-6 prodotta dalle cellule stromali del midollo che fa crescere le plasmacellule attraverso i recettori per l'IL-6 sulle plasmacellule, ma esistono anche modelli più avanzati di malattia, un modello appunto autocrino, dove è la stessa plasmacellula che produce IL-6 e che serve alla proprio proliferazione. Ma sicuramente oltre a IL-6 tanti altri fattori intervengono nella proliferazione delle plasmacellule e qui vedete rappresentato (slide) il TNFα che viene prodotto prevalentemente a seguito di un contatto tra la plasmacellula e la cellula stromale. (slide) Vedete qui c'è un contatto diretto, c'è bisogno proprio di un contatto fisico. Ecco perché le plasmacellule proliferano nel midollo osseo, perché lì trovano le cellule stromali al contatto delle quali poi si innescano tutta una serie di momenti di attivazione citochinica e vedete in particolare qua (slide) la cellula stromale che produce IL-6 ma questo determina anche la liberazione di TNFα da parte delle plasmacellule, il quale TNFα poi reagisce sulla cellula stromale inducendo tutta una serie di attività a cascata che provocano una ulteriore produzione di IL-6. Quindi estremamente importante questo contatto che esiste tra la cellula mielomatosa e le cellule stromali. Vi dicevo che la caratteristica laboratoristica di questa malattia è la produzione di una componente monoclonale al protidogramma, la cosiddetta Gammapatia monoclonale. “Gammapatia” perché appunto questo picco monoclonale è più spesso presente presente in zona γ. Voi sapete che nel protidogramma ci sono le albumine, le α1, α2, le β e le γ, a seconda appunto della zona in cui le proteine si distribuiscono. In genere, ma non sempre, nel mieloma multiplo queste proteine vanno a finire nella zona γ e quindi si chiama Gammapatia. Però non tutte le gammapatie sono mieloma multiplo, perché esistono diverse condizioni. Una è la cosiddetta Gammapatia Monoclonale di Incerto Significato (MGUS) che è peraltro la più frequente, quella che una volta si chiamava gammapatia monoclonale benigna perché appunto non è una patologia ma è una condizione nella quale noi troviamo al protidogramma questo aumento delle gammaglobuline monoclonali. Una volta quindi si chiamava Gammapatia monoclonale benigna, poi però visto che questa condizione può progredire verso un mieloma multiplo le assicurazioni americane sono riuscite a far cambiare questo nome da Gammapatia monoclonale benigna a Gammapatia monoclonale di incerto significato. “Incerto significato” vuol dire che potrebbe essere benigna ma potrebbe anche evolvere verso una forma maligna. Quindi da questa condizione che è una condizione assolutamente benigna nel senso che non si accompagna a nessuna alterazione dell'organismo poi ci sono tutte le altre forme di mieloma che può essere un mieloma cosiddetto “smouldering”, cioè un mieloma indolente, non attivo oppure il cosiddetto plasmacitoma solitario. Nel plasmacitoma solitario c'è generalmente una localizzazione plasmacellulare a livello usualmente di un osso, ma anche in altre parti del corpo e non sempre però c'è la componente monoclonale in periferia. E poi c'è il classico mieloma multiplo che può dare anche localizzazioni extramidollari o infine nella fase più avanzata la cosiddetta leucemia plasmacellulare, cioè queste plasmacellule che sono diventate tanto maligne da perdere la necessità di un contatto con lo stroma midollare e quindi crescono autonomamente e sono quelle plasmacellule che producono loro stesse per esempio IL-6 e quindi possono autoalimentarsi. Questo un po' nella fase finale della malattia mielomatosa. (slide) Quindi questo è il quadro elettroforetico tipico di un mieloma multiplo, vedete l'aspetto cosiddetto “a corna”, o in maniera un po' più elegante “a orecchio di coniglio” dove esistono appunto l'albumina e le γ-globuline che hanno in pratica la stessa morfologia. Questo ci dice che esiste una componente monoclonale a livello della zona γ. Ma per sapere quale tipo di immunoglobulina è interessata bisogna fare un altro esame che si chiama immunoelettroforesi. Nell'immunoelettroforesi vengono testate le tre principali catene pesanti delle immunoglobuline (IgG, IgA e IgM) e le catene leggere κ e λ. (slide) Come vedete in questo caso c'è una netta monoclonalità per le catene pesanti G e per le catene leggere κ. Quindi in questo caso si tratta di una Gammapatia monoclonale IgG-κ, cioè sono plasmacellule che producono tutte un solo tipo di catena pesante che è la IgG e un solo tipo di catena leggera che è la κ. Questo è importante perché è l'unico esame che ci dimostra con certezza la monoclonalità perché l'elettroforesi delle proteine qui (slide) è abbastanza evidente ma non sempre è così evidente, a volte il picco monoclonale ha una base un pochettino più larga, come succede per esempio nelle patologie in cui c'è una eccessiva produzione di immunoglobuline come per esempio nelle cirrosi epatiche in cui c'è una eccessiva produzione di immunoglobuline che però non sono monoclonali, sono policlonali e quindi si distribuiscono su un range più ampio di punto isoelettrico e di conseguenza danno un aumento delle gammaglobuline di tipo policlonale appunto con una base molto più larga. Per cui l'esame che ci dice con certezza che si tratta di una produzione monoclonale di immunoglobuline è appunto la immunofissazione elettroforetica. Quindi fondamentalmente quando ci troviamo di fronte ad un paziente che ha un protidogramma con un picco monoclonale in zona γ o più raramente in zona β, fondamentalmente possiamo avere 3 condizioni: − Gammapatia monoclonale di incerto significato; − Mieloma asintomatico, il cosiddetto “smouldering” myeloma; − Mieloma multiplo. Queste sono le condizioni più frequenti. Poi c'è qualche altra condizione che vedremo successivamente, ma fondamentalmente queste tre condizioni sono quelle da tenere in considerazione. È importante definire queste condizioni. Si definisce come MGUS, cioè Gammapatia Monoclonale di Incerto Significato, quella condizione in cui la componente la componente sierica di proteine è <3 g/dL o <30 g/L, una condizione nella quale la percentuale di plasmacellule monoclonali nel midollo è <10%, non ci deve essere evidenza di malattie linfoproliferative le quali si possono accompagnare ad una componente monoclonale. Molte malattie proliferative del sistema linfoide, proprio perché determinano la iperplasia della linea linfoide, si possono accompagnare ad una Gammapatia monoclonale generalmente appunto in zona γ e quindi bisogna escludere che uno abbia un linfoma per esempio. Soprattutto la cosa importante è che non ci deve essere una compromissione d'organo o di tessuto per definire la Gammapatia monoclonale di incerto significato. L'altra condizione possibile è un mieloma che però è asintomatico. In questo caso o la componente monoclonale è superiore o uguale a 3g e/o la percentuale di plasmacellule nel midollo è superiore o uguale al 10%, ma ancora una volta non ci devono essere compromissioni di organo o di tessuto. Viceversa la caratteristica che ci permette di fare diagnosi di mieloma è proprio la presenza di una compromissione d'organo o di tessuto in presenza di una paraproteina, cioè di una componente monoclonale. Badate che qui (slide) non c'è scritto quanto deve essere, se deve essere inferiore a 3 g o superiore a 3 g perché di qualunque entità sia la paraproteina e di qualunque entità sia la presenza di plasmacellule nel midollo l'importante è che ci siano queste compromissioni d'organo. Quindi la diagnosi di mieloma va fatta sulla base di queste compromissioni d'organo e queste compromissioni d'organo sono definite con l'acronimo CRAB. C= aumento della Calcemia R= compromissione Renale A= Anemia B= lesioni ossee (Bone marrow) Voi sapete che in inglese “Crab” significa “granchio”. Per fare una diagnosi di mieloma ci vogliono queste condizioni (compromissioni di organo) in presenza di una componente monoclonale e di una infiltrazione plasmacellulare midollare di qualunque entità. Quindi su un piano fisiopatogenetico il mieloma multiplo non è altro che l'evoluzione di un'alterazione del linfocita che si manifesta con una iperplasia delle plasmacellule, ma probabilmente il difetto iniziale nasce nei linfociti, poi questi linfociti riescono a maturare fino allo stadio di plasmacellula e danno quella condizione che abbiamo definito Gammapatia monoclonale di incerto significato. Tutto questo può rimanere a questo livello, stabile, oppure può evolvere verso un mieloma. Quindi inizialmente verso un mieloma “smouldering”, poi verso un mieloma intramidollare, poi verso un mieloma extramidollare e tutto questo dipende non soltanto dalle alterazioni citogenetiche e molecolari che intervengono in queste plasmacellule di MGUS, ma anche da tutta una serie di eventi che intervengono nel microambiente midollare come per esempio: − produzione di IL-6; − neoangiogenesi; − l'aumentata produzione di citochine che distruggono poi l'osso; Anche le plasmacellule del MGUS presentano delle alterazioni cromosomiche che inducono instabilità genomica e appunto non solo questo, ma soprattutto delle modifiche del microambiente midollare, sono tutte condizioni che favoriscono lo sviluppo del mieloma franco dove ovviamente intervengono tutta una serie di alterazioni citogenetiche e molecolari per esempio i RASK, i RAS etc che favoriscono la proliferazione di queste plasmacellule. Però vi dicevo che quando noi vediamo un paziente con una Gammapatia monoclonale non sempre si tratta di un mieloma, anzi più spesso si tratta di un MGUS. (slide) Per esempio una casistica di una istituzione americana ha valutato tutti i pazienti che venivano osservati per una componente monoclonale e la stragrande maggioranza di questi pazienti avevano un MGUS, cioè una Gammapatia monoclonale di incerto significato. Soltanto una piccola quota di questi pazienti aveva un mieloma multiplo, una piccola quota aveva uno smouldering myeloma, una quota non significativa aveva un'altra patologia chiamata amiloidosi di cui parleremo successivamente e una piccola quota aveva un'altra malattia linfoproliferativa. Quindi per studiare la componente monoclonale la cosa più importante è fare l'elettroforesi delle proteine sia sieriche che urinarie e quindi dosare questo picco monoclonale in grammi, fare un dosaggio nefelometrico delle immunoglobuline IgG, IgA e IgM (queste sono soltanto le catene pesanti), fare l'immunofissazione che vi ho fatto vedere prima sia sul siero che sulle urine e misurare appunto sulle urine la quantità di catene leggere. Le immunoglobuline infatti sono formate da catene pesanti e catene leggere, le catene pesanti non passano il filtro glomerulare, mentre le catene leggere passano il filtro glomerulare. Quindi la misurazione delle catene leggere nelle urine ci dà la misura di quante immunoglobuline vengono prodotte nell'ambito delle catene leggere. Questo una volta si faceva con un metodo al calore che veniva chiamata “proteinuria di BenceJones”, cioè scaldando queste urine in realtà si venivano a creare dei flocculati, degli aggregati proteici che definivano la cosiddetta proteinuria di Bence-Jones. Adesso non si usa più fare la proteinuria di Bence-Jones, si usa una quantificazione un pochettino più accurata ma il concetto è sempre lo stesso, cioè valutare nelle urine la presenza di catene leggere monoclonali. Bisogna fare anche degli altri esami importanti, bisogna fare l'emocromo, valutare la calcemia, perché la calcemia può essere aumentata in questi pazienti in quanto c'è una distruzione ossea, valutare la cosiddetta β2-microglobulina sierica che ha un valore prognostico perché la β2microglobulina è un antigene presente sulla membrana di queste plasmacellule che può essere liberato in circolo e anzi tanto maggiore è il turnover plasmacellulare quanta più β2-microglobulina si libera in circolo e quindi viene dosata nel plasma. È importante misurare la creatinina, l'azotemia che sono espressione appunto della funzionalità renale perché vi dicevo che i reni sono spesso coinvolti nel mieloma. È importante misurare anche la proteina C reattiva perché la proteina C è un po' lo specchio dell'IL-6. IL-6 non fa altro che stimolare il fegato a produrre proteina C, per cui se noi abbiamo tanta proteina C questo è un indice che c'è tanta IL-6 e come visto prima l’IL-6 è il fattore di crescita più importante per le plasmacellule mieloma tose: dal momento che dosare IL-6 è piuttosto indaginoso, non è un esame di routine, allora noi dosiamo la PCR. La PCR può essere aumentata per tanti altri motivi però sicuramente i pazienti che hanno la PCR aumentata hanno tanta IL-6, poi può essere che la PCR è aumentata perché hanno una flogosi anzi sicuramente perché c'è una flogosi, anche nelle flogosi aumenta IL-6 e aumenta la PCR, però è una valutazione indiretta di quanta IL-6 c'è in giro. Quindi la Gammapatia monoclonale di incerto significato si chiama così perché una piccola quota di questi pazienti può evolvere verso un mieloma franco e relativamente di recente è stata fatta una valutazione appunto della percentuale di questi pazienti che evolvono verso il mieloma ed è stato visto che su 241 pazienti che sono stati seguiti per un follow-up mediano di 13 anni c'è stato un incremento della componente M del 10%, morti per cause non correlate 57% (morti di vecchiaia, di altri motivi), progressione 27%. Progressione in che cosa? La maggior parte ha sviluppato un mieloma, qualcun altro ha sviluppato una macroglobulinemia di Waldenstrom che è un'altra malattia linfoproliferativa un po' a cavallo tra i linfomi e i mielomi, una piccola quota ha sviluppato l'amiloidosi e altri hanno sviluppato altri disordini linfoproliferativi. Quindi andando a valutare il rischio di progressione di questi pazienti si è calcolato che il rischio di progressione è di circa 1% l'anno ed è cumulativo, cioè il paziente con MGUS oggi ha una probabilità di 1% di sviluppare un mieloma, tra un anno avrà 2%, tra due anni avrà il 3% e così via. (slide) Questa è la curva di progressione di MGUS verso il mieloma multiplo. Come vedete appunto a 20 anni circa il 20% di questi pazienti ha avuto un rischio di progressione verso il mieloma multiplo. Molto più alta ma non moltissimo più alta è la curva di progressione del cosiddetto mieloma smouldering. Ricordiamo che il mieloma smouldering è una condizione nella quale o il picco monoclonale è abbastanza alto, cioè più di 3 g/dL oppure la quantità di plasmacellule midollari è superiore al 10% o ambedue le cose, però questi pazienti non hanno i segni CRAB, cioè non hanno alcuna compromissione d'organo o di tessuto. In questi pazienti noi facciamo diagnosi di mieloma smouldering, cioè hanno una grossa componente monoclonale, hanno un grosso infiltrato plasmacellulare nel midollo, ma non hanno nessuna compromissione d'organo. (slide) Questi pazienti vedete qui hanno una elevata possibilità di trasformazione verso il mieloma però considerati a 5 anni, soltanto la metà di questi pazienti è andata incontro ad evoluzione a mieloma, quindi c'è almeno un'altra metà di pazienti che è rimasta stabile. Quindi quello che è importante nel trattamento del mieloma, perché va trattato solo il mieloma sintomatico non il mieloma smouldering, almeno per il momento il mieloma è una malattia che ancora non riusciamo a far guarire almeno nella stragrande maggioranza dei casi, quindi i pazienti vanno trattati solo se hanno un motivo per essere trattati. Se un paziente ha soltanto un mieloma smouldering non va trattato e come vedete la probabilità che un mieloma smouldering si trasformi in un mieloma vero e proprio è contenuta, cioè al 50%, non è altissima a 5 anni e visto che si tratta di pazienti anziani molto spesso, un paziente che ha 75 anni per esempio a cui offriamo almeno altri 5 anni privi di progressione di malattia non sembra una cosa tanto da buttare via. Chiaramente anche il mieloma smouldering ha dei gruppi di rischio, ovvero se un paziente ha più del 10% di plasmacellule e una componente monoclonale più di 3 g/dL sicuramente è più a rischio di un paziente che ha o l'una o l'altra condizione. (slide) Qui vedete la curva di progressione. Questi sono i pazienti che hanno più del 10% di plasmacellule e più di 3g di proteina. Il Gruppo 3 invece riguarda quelli che hanno meno del 10% di plasmacellule e una componente monoclonale più di 3 g/dL. Quindi ricapitolando quello che abbiamo visto prima abbiamo una condizione di Gammapatia monoclonale di incerto significato dove c'è vedete (slide) questo “picchetto” monoclonale che è presente in zona γ e qui abbiamo meno del 10% di plasmacellule, sicuramente il paziente è asintomatico e non ha nessun danno d'organo. In questo caso bisogna solo osservarlo. Il mieloma smouldering dove sicuramente la componente monoclonale è maggiore, il numero di plasmacellule può essere superiore al 10%, ma anche qui è asintomatico e nessun danno d'organo e anche qui soltanto osservazione. Viceversa il paziente con mieloma, a prescindere dall'entità della componente monoclonale, ma con danno d'organo in questo caso è richiesta la terapia. In tutti i libri troverete questa stadiazione del mieloma multiplo che è la stadiazione di Durie e Salmon che risale circa al '68 e che è ancora valida e dove il mieloma viene stadiato in 3 stadi a seconda di alcune caratteristiche che sono anche un po' difficili da ricordare però fondamentalmente: − Stadio I: paziente che non è anemico, che non è ipercalcemico, che non ha lesioni osteolitiche e che ha una bassa quantità di componente monoclonale. IgG<5g/dL IgA<3g/dL, catene leggere delle urine <4g/24h. − Stadio II: stadio intermedio tra I e III. − Stadio III: paziente anemico, ipercalcemico, con molte lesioni scheletriche e il paziente che ha una elevata quantità di componente monoclonale nel siero e nelle urine. Poi questi pazienti vengono distinti in A e B a seconda che hanno una creatinina<2mg/dL (A) o una creatinina >2mg/dL (B). Quindi fondamentalmente questa stadiazione prevede 3 stadi: nel primo stadio quindi assenza di anemia, assenza di ipercalcemia, scheletro normale, bassa quantità di immunoglobuline; il terzo stadio è il contrario cioè anemia, ipercalcemia, numerose lesioni scheletriche, alta quantità di immunoglobuline. Lo stadio due è uno stadio intermedio tra queste due condizioni. Però vi dicevo che questo è anche un po' difficile da ricordare, quindi di recente sono state proposte nuove classificazioni e tra le nuove classificazioni quella che più è stata accreditata anche per la sua semplicità mnemonica è questa nuova classificazione definita “International Staging System” dove fondamentalmente sono stati utilizzati 2 criteri: − della β2-microglobulina che come dicevo è espressione del turnover cellulare, quindi espressione di quanto questa malattia cresce; − albumina, espressione della risposta dell'organismo alla malattia neoplastica. Il paziente che è ipoalbuminemico (e questo vale un po' per tutte le malattie neoplastiche) è un paziente che ha una forte compromissione della capacità protido-sintetica del fegato e quindi è un paziente in cui la malattia neoplastica ha presto il sopravvento sulla sua normale omeostasi. Vengono definiti: − Stadio I: pazienti che hanno una bassa β2-microglobulina, cioè <3,5mg/L e una alta albumina, >3,5g/dL. I numeri sono messi apposta per essere facilmente ricordati, 3.5 per ambedue. − Stadio II: intermedio tra I e III. − Stadio III: caratterizzato dalla β2-microglobulina >5,5. Vi ricordo però che la β2-microglobulina risente molto della escrezione renale, quindi nello stadio III ci stanno non solo i pazienti che hanno una β2-microglobulina aumentata per l'elevato turnover cellulare delle plasmacellule mielomatose, ma anche quelli che hanno una elevata β2microglobulina perché c'è una compromissione renale che riduce l'escrezione di β2-microglobulina. La β2-microglobulina è un po' come la creatinina, cioè viene eliminata per via renale e quindi un danno renale determina un incremento plasmatico di questa proteina. Questo non fa altro che sottolineare l'importanza prognostica negativa di un danno renale nei pazienti con malattia mielomatosa. Infatti se voi vedete la sopravvivenza mediana di questi pazienti, in stadio I, è una sopravvivenza mediana di 62 mesi, in stadio II 44 mesi, in stadio III 29 mesi soltanto. Quindi i primi pazienti hanno una sopravvivenza mediana di 5 anni grossomodo, i secondi hanno una sopravvivenza mediana di poco più di 2 anni. Lo stadio II è lo stadio intermedio tra I e III e fondamentalmente ci sono 2 categorie di stadio II: − stadio II con β2-microglobulina bassa, ma bassa albumina: − stadio II con β2-microglobulina tra 3.5 e 5.5 senza considerare i livelli di albumina. Per la prognosi di questi pazienti non basta valutare lo stadio di malattia, ma è molto importante come abbiamo visto per la leucemia linfatica cronica la citogenetica. In quasi tutte le malattie ematologiche ormai la citogenetica ha un ruolo importantissimo nel predire la prognosi di queste malattie perché la citogenetica fondamentalmente indica quelle alterazioni che poi sono probabilmente in qualche modo responsabili della progressione della malattia, indica la “malignità” del clone neoplastico. Tante più alterazioni molecolari e citogenetiche ci sono, tanto più “maligno” è questo clone neoplastico, tanto più è resistente al trattamento. Però nel mieloma c'è una peculiarità che occorre sottolineare. (slide) Se voi vedete in questa curva di sopravvivenza quelli che stanno al top della curva di sopravvivenza sono i pazienti che non hanno alterazioni citogenetiche né in FISH, né in citogenetica convenzionale. Sapete che per fare la citogenetica occorre prendere del materiale biologico, stimolare la mitosi di queste cellule e bloccare poi queste cellule in metafase. Succede che molte cellule neoplastiche che hanno un basso indice proliferativo non vanno in mitosi, questo succede ad es. per la leucemia linfatica cronica ma anche per il mieloma. Voi considerate che l'indice proliferativo del mieloma è intorno a 1-2%, cioè se noi prendiamo tutte le cellule mielomatose e vediamo quante di queste cellule sono in mitosi ne troviamo da 1% al 2%, quindi è una piccola quota di cellule che sono in mitosi. Anche con le tecniche di stimolazione della mitosi solo una quota di queste cellule va incontro alla mitosi e quindi può essere osservata in citogenetica convenzionale perché la citogenetica vede le cellule che sono andate in mitosi, blocca la mitosi e poi vede i cromosomi. Se le cellule non vanno in mitosi la citogenetica fallisce, non riesce a vedere le alterazioni cromosomiche e allora la metodica che si utilizza è la cosiddetta FISH (Fluorescent In Situ Hibrydization) cioè si marcano alcuni cromosomi e si vede se c'è la traslocazione. Se noi facciamo l'esempio di una traslocazione, come quella della mieloide cronica, la (9:22) e marchiamo con due colori, per esempio con una sonda verde e una sonda rossa i due cromosomi 9 e 22: se questi due cromosomi hanno una traslocazione e quindi si fondono parti di cromosomi, rosso più verde dà giallo e quindi noi invece di vedere un segnale rosso e un segnale verde vedremo un segnale giallo e questo ci dice che c'è la traslocazione, per quella condizione che siamo andati a ricercare ovviamente. Il limite della FISH è che ci permette di andare ad evidenziare le alterazioni cromosomiche anche in cellule che non si dividono ma limitatamente alle alterazioni che andiamo a ricercare, mentre la citogenetica convenzionale possiamo vedere tutti i cromosomi. I pazienti che hanno le alterazioni come la delezione del 13 né in FISH né in citogenetica convenzionale sono i pazienti che vanno meglio. I pazienti che hanno la delezione del 13 soltanto in FISH e non in citogenetica convenzionale vanno molto meglio rispetto ai pazienti che hanno l'alterazione in citogenetica convenzionale e in FISH, questo perché come dicevamo prima la FISH è in grado di valutare le alterazioni citogenetiche nelle cellule che non si dividono mentre la citogenetica convenzionale è in grado di valutarlo soltanto nelle cellule che si dividono. Avere una alterazione del 13 in citogenetica convenzionale significa non solo avere la delezione del 13 e quindi queste anomalia cromosomica, ma averla in cellule che proliferano, che si dividono. Viceversa averla soltanto in FISH, cioè soltanto in cellule che non si dividono e non riuscire a vederla in citogenetica convenzionale significa che queste cellule sì hanno l'alterazione citogenetica, ma non sono cellule che crescono rapidamente, che proliferano. Quindi in questo caso è anche la metodica con cui noi rileviamo le alterazioni citogenetiche ad avere un peso sulla prognosi. Nel caso specifico, una alterazione citogenetica come la delezione di 13 valutata in citogenetica convenzionale ha un peso fortemente negativo, mentre questa alterazione citogenetica valutata in FISH cioè in cellule che non si dividono ha un peso di gran lunga migliore. Tutto questo per introdurre il discorso che nel mieloma le anomalie citogenetiche sono molto frequenti. Nel mieloma fondamentalmente distinguiamo 2 tipi di anomalie citogenetiche: − i pazienti che sono iperdiploidi che sono circa la metà e che hanno questa iperdiploidia per trisomie dei cromosomi 3,5,7,9,11,15,19 curiosamente sono tutti cromosomi dispari. Quindi la metà di questi pazienti sono iperdiploidi perché hanno una o più di queste trisomie. − Un'altra metà di pazienti ha una delezione del 13, di cui parlavamo prima oppure hanno una acquisizione del cromosoma 1 oppure hanno una anomalia del cromosoma 14. Ritorna qui il famoso cromosoma 14 in cui stanno i geni delle immunoglobuline e parlando di plasmacellule che producono immunoglobuline sembra il minimo che il cromosoma 14 sia interessato nelle alterazioni citogenetiche. Quindi grossomodo possiamo distinguere 2 tipi: i pazienti con cariotipo iperdiploide e i pazienti con cariotipo non iperdiploide e grossomodo sono 50 e 50. La cosa interessante è che se noi prendiamo le alterazioni che poi hanno un significato prognostico negativo tipo la delezione del 13, questa è molto più frequente nel cariotipo non iperdiploide. Lo stesso per la traslocazione 14q32, cioè l'interessamento del cromosoma 14. Questo fa sì che il cariotipo iperdiploide abbia una prognosi migliore rispetto al cariotipo non iperdiploide. Tutto questo fondamentalmente dipende dal fatto che le alterazioni citogenetiche sfavorevoli come la (4;14), la (14;16), tutta una serie di alterazioni che interessano il 14 sono più frequenti nel cariotipo non iperdiploide rispetto a quello iperdiploide. Detto questo sulla biologia della malattia vediamo qual è la sintomatologia, la clinica del paziente con mieloma. Intanto c'è una piccola quota, un 30% di pazienti in cui abbiamo un riscontro asintomatico, casuale, sono pazienti che fanno il protidogramma per un altro motivo e nella maggior parte dei casi si tratta di MGUS, ma in qualche caso possiamo anche beccare qualche mieloma. Il sintomo principale, il sintomo cardine del paziente con mieloma sono i dolori ossei. Purtroppo questi dolori ossei vengono spesso misconosciuti o vengono mal interpretati perché siccome si tratta di pazienti anziani e i pazienti anziani si lamentano sempre di dolori articolari più che ossei e allora vengono scambiati come patologia artrosica, patologia degenerativa e spesso si tratta di pazienti che fanno due o tre visite dall'ortopedico prima che l'ortopedico si decida a prescrivere un protidogramma e poi mandare il paziente dall'ematologo. Quindi se vedete un paziente anziano con dolori ossei, invece di mandarlo dall'ortopedico per prima cosa fategli fare un protidogramma per vedere se al protidogramma troverete un’alterazione che possa indicare appunto un sospetto di mieloma. Poi sono pazienti che possono essere anemici, sono pazienti che possono avere infezioni recidivanti, insufficienza renale e sindrome ipercalcemica. Qual è la genesi di questi sintomi? Intanto la cellula mielomatosa abbiamo visto che cresce e prolifera a livello midollare e quindi va a sostituirsi nel midollo alle normali popolazioni midollari, questa condizione induce deficit di produzione di globuli rossi, quindi anemia, deficit di produzione di globuli bianchi, quindi granulocitopenia e deficit di produzione delle piastrine e quindi piastrinopenia. La piastrinopenia è aggravata nella sua manifestazione emorragica dal fatto che la componente monoclonale che viene prodotta da queste plasmacellule può anche interferire con la membrana piastrinica, quindi oltre ad esserci una piastrinopenia c'è anche una piastrinopatia, cioè una minore funzionalità delle piastrine. Anche la granulocitopenia che sapete è responsabile delle infezioni viene potenziata in questo rischio infettivo dal fatto che producendosi un solo tipo di immunoglobulina, per esempio IgG monoclonale, questo determina la retroinibizione delle altre immunoglobuline, sia delle normali immunoglobuline G, sia delle IgA e delle IgM, quindi determina una condizione di ipogammaglobulinemia delle γ−globuline normali che facilita le infezioni. Quindi i motivi delle infezioni sono da un lato la granulocitopenia, dall'altro la ipogammaglobulinemia delle normali immunoglobuline. C'è una minore sintesi di immunoglobuline normali e quindi aumenta la suscettibilità alle infezioni, ma l'aspetto più importante è che produce una componente monoclonale che può dare intanto emodiluizione, soprattutto quando si tratta di IgA o di IgM, le IgA sono dimeriche, le IgM sono pentameriche e possono quando sono in eccesso dare una sindrome da iperviscosità e da emodiluizione. Quindi aumento della viscosità ematica che può essere moderata o grave, vi dicevo possibilità di interferenza con la membrana piastrinica, ma anche con i fattori della coagulazione. Queste immunoglobuline possono agire nei confronti di alcuni fattori della coagulazione, per esempio nei confronti del fattore VIII, il fattore V. Si possono depositare a livello dei parenchimi queste immunoglobuline e l'effetto più frequente è il danno glomerulare, soprattutto tubulare a carico dei reni e quindi l'insufficienza renale oppure possono anche depositarsi in altri organi e dare quella che viene definita amiloidosi secondaria e poi vedremo un po' cos'è la amiloidosi. Oppure queste immunoglobuline possono precipitare al freddo e dare una condizione di tipo crioglobulinemico. Inoltre la cellula mielomatosa produce dei fattori che attivano gli osteoclasti. L'aumento dell'attività degli osteoclasti determina un aumento del riassorbimento della matrice ossea e tutto questo provoca le lesioni osteolitiche tipiche appunto del mieloma, con possibilità anche di fratture patologiche, non solo lesioni osteolitiche ma anche una grave osteoporosi che assieme appunto a queste lesioni osteolitiche è responsabile della sintomatologia più frequente che è quella del dolore osseo. Accanto a questo, questa aumentata attività osteoclastica quindi aumentato riassorbimento osseo può provocare una ipercalcemia che può essere: − ipercalcemia lieve quando la calcemia è <12g e in questo caso non c'è usualmente sintomatologia; − può essere moderata e in questo caso può esserci una sintomatologia abbastanza sfumata, − il paziente soltanto lamenta una astenia e una poliuria; − ipercalcemia grave e in questo caso si può avere una cosiddetta sindrome da ipercalcemia con dolore addominale, nausea, astenia, poliuria, può portare al coma e addirittura alla morte. Allora è importante nel paziente con mieloma andare a definire se il paziente se ha una sindrome ipercalcemica oppure no. Ricordatevi che quando il laboratorio ci dà il valore della calcemia noi dobbiamo correggere questo valore di calcemia con un fattore correttivo e questo perché il calcio diventa tossico quando è libero. Normalmente il calcio è infatti legato all'albumina, il calcio che non riesce a legarsi all'albumina diventa tossico per il sistema nervoso sia centrale che periferico e quindi dà quei sintomi che abbiamo visto, astenia, dolori addominali, nausea, il coma quando c'è troppo calcio fino alla morte. E quindi se è il calcio libero quello che dà la sintomatologia è chiaro che se noi abbassiamo l'albumina c'è meno possibilità di legare il calcio e quindi c'è più calcio libero e pertanto maggiore sintomatologia. Allora quando noi andiamo a valutare la sindrome ipercalcemica non dobbiamo considerare il valore che ci dà il laboratorio del calcio, ma dobbiamo correggere questo valore per l'albumina. Allora la formula semplice che il prof utilizza è di considerare l'albumina normale come come 4. Quindi valore della calcemia, per esempio 12mg + 4 – il valore dell'albumina che ha il paziente. Quindi se il paziente ha un valore normale di albumina sarà il valore della calcemia + 4 – 4. 12+4-4=12, lo stesso valore che ha dato il laboratorio. Ma se il paziente ha 2 di albumina facciamo 12+4-2=14, quindi andiamo nel range di una ipercalcemia, anche se abbiamo un valore quasi normale di calcio. Domanda di una collega: “Le alterazioni dell'albumina dipendono da una disfunzionalità epatica?” Risposta: “Sì. Dipende da tante condizioni però i pazienti neoplastici hanno spesso una compromissione del fegato che non produce più albumina in maniera sufficiente, poi può essere anche che quello ha una sindrome nefrosica, per esempio nel mieloma è frequente la sindrome nefrosica per compromissione renale e allora l'albumina non è che non viene prodotta ma viene persa con le urine e allora in quel caso a prescindere dalla capacità protido-sintetica del fegato il paziente ha meno albumina e quindi questo può favorire la sindrome ipercalcemica. La sindrome ipercalcemica è quindi dovuta alla distruzione dell'osso che provoca dei sintomi generali, quindi astenia, obnubilamento del sensorio, nausea e vomito, ma siccome il calcio viene eliminato con le urine determina una ipercalciuria. L'ipercalciuria provoca un forte aumento della diuresi osmotica e quindi una poliuria e dunque una disidratazione che va a peggiorare l'ipercalcemia e favorisce l'insufficienza renale. Quindi l'insufficienza renale dipende da tante cause nel mieloma: − dal fatto che il calcio è in qualche modo tossico, perché quando c'è una eccessiva presenza di calcio nel tubulo questo provoca una nefrocalcinosi e favorisce l'insufficienza renale; − il calcio favorisce la diuresi osmotica e quindi la poliuria e dunque la disidratazione e questo favorisce l'insufficienza renale; − l'insufficienza renale viene anche favorita ovviamente dalla presenza di catene leggere nelle urine. Accanto alla sindrome da ipercalcemia esiste anche la sindrome da iperviscosità che dipende appunto dal fatto che quando c'è una componente monoclonale molto rappresentata, soprattutto se queste immunoglobuline sono dimeriche o pentameriche, provocano un significativo aumento della viscosità ematica e quindi un rallentamento del microcircolo e questo rallentamento del microcircolo provoca ovviamente un aumento della resistenza e quindi un aumento del lavoro cardiaco. Un rallentamento del microcircolo nel SNC e quindi disturbi neurologici da ipoperfusione cerebrale e un rallentamento del microcircolo periferico, quindi con una insufficienza periferica. Vi dicevo, è molto importante il fatto che queste immunoglobuline possono anche precipitare, soprattutto le catene leggere delle immunoglobuline tendono a precipitare. Tutto questo permette la formazione della cosiddetta sostanza amiloide. La sostanza amiloide non è altro che un tessuto amorfo che è conseguenza della precipitazione delle catene leggere a livello dei vari organi. Questo può provocare una organomegalia senza deficit funzionale, soprattutto epatomegalia, oppure tipico dell'amiloidosi è la sindrome del tunnel carpale. Io vedo pazienti che si fanno il tunnel carpale bilaterale e nessuno gli chiede un protidogramma!! Se un paziente ha una sindrome da tunnel carpale bilaterale, la prima cosa da chiedere è il protidogramma perché può essere espressione di una amiloidosi. Oppure se questa sostanza amiloide depositata a livello dei vari organi provoca un difetto funzionale possiamo avere insufficienza renale, insufficienza cardiaca, neuropatia, malassorbimento, tutti i sintomi della amiloidosi. Nel mieloma ci può essere una sintomatologia neurologica che può dipendere da diverse condizioni: abbiamo visto come ci può essere infiltrazione di componente amiloide in vari organi, compreso il tessuto nervoso periferico e in questo caso c'è una polineuropatia periferica. Ma dal momento che la malattia cresce nel midollo osseo → il midollo osseo sta soprattutto nelle vertebre → quindi dalle vertebre la malattia può crescere e dare compressione del midollo spinale, quindi può esserci presenza di masse e manicotti a livello del rachide che determinano una compressione o un crollo vertebrale e quindi una sindrome spinale per compressione del midollo spinale. (slide) Qui sono presenti alcuni quadri radiologici di lesione osteolitica. Qui vedete un cranio tutto “bucherellato”, qui vedete una vertebra con tutte queste lesioni osteolitiche, qui vedete una frattura di un omero, una frattura cosiddetta “patologica”. Questo perché appunto le lesioni osteolitiche sono molto frequenti e sono frequenti soprattutto in quelle parti dove si trova il midollo osseo perché abbiamo visto che sono le plasmacellule che producono delle sostanze che attivano poi gli osteoclasti i quali determinano il riassorbimento osseo. Le plasmacellule stanno nel midollo osseo, il midollo osseo sta soprattutto nello scheletro assiale nell'adulto e quindi è abbastanza normale che la maggior parte delle lesioni osteolitiche intervengano nel cranio, nella colonna e nel bacino. Come si evidenziano queste lesioni osteolitiche? Le lesioni osteolitiche si evidenziano con la radiologia tradizionale, con la TAC, con la RM e con la PET. Si possono evidenziare anche con una scintigrafia particolare che si chiama Sestamibi ma non con la scintigrafia ossea tradizionale con il tecnezio. Quando in reparto il prof e il suo staff richiedono la radiografia del cranio, della colonna e del bacino con la radiologia tradizionale, quasi sempre ci viene richiesto “perché invece di fare tutte queste radiografie, non facciamo solo una scintigrafia ossea per avere un quadro più completo di tutto il corpo anche per sottoporre il paziente a meno radiografie?”. Questo discorso non è applicabile nel mieloma perché la scintigrafia con tecnezio si basa sul fatto che il tecnezio viene captato dagli osteoblasti. Il tecnezio va a localizzarsi sugli osteoblasti, cioè su quelle cellule che tentano di riparare la lesione ossea e questo va bene per tutte le lesioni osteolitiche dei tumori solidi dove anche alla radiografia tradizionale si vede la lesione osteolitica con l'orletto riparativo, cioè la lesione che cerca di essere riparata dagli osteoblasti. Nel mieloma invece se voi vedete una radiografia tradizionale non si vede l'orletto riparativo proprio perché non ci sono osteoblasti che riparano, perché nel mieloma la patogenesi della lesione osteolitica non è soltanto l'attivazione osteoclastica, ma è anche l'inibizione degli osteoblasti. Allora se noi facciamo una scintigrafia con tecnezio può succedere che essendoci pochi osteoblasti che riparano questa lesione non viene captato il tecnezio in quella lesione osteolitica e magari può succedere che la scintigrafia è muta nonostante ci sia la lesione osteolitica. Quindi per andare a valutare le lesioni osteolitiche del mieloma non è utile la scintigrafia ossea con tecnezio, ma sono utili altre metodiche. Non è detto che la scintigrafia ossea non sia positiva, anzi il modo migliore per andare a vedere le lesioni osteolitiche delle coste è proprio la scintigrafia ossea col tecnezio! Questo però ripetiamo è limitato a quelle condizioni in cui c'è anche un'attivazione osteoblastica. Se non c'è la attivazione osteoblastica la scintigrafia con tecnezio diventa muta. Quindi la scintigrafia con tecnezio non è l'esame principe nei pazienti con lesioni osteolitiche da mieloma. (slide) Qui vedete una vertebra del tutto erosa da un processo infiltrativo. Qui vedete invece una vertebra con questa localizzazione mielomatosa che pigia sul canale midollare, sul midollo spinale e che dà una sindrome da compressione radicolare. Queste sono sindromi molto importanti perché sono pazienti che dal momento in cui si paralizzano, (perché più spesso si paralizzano gli arti inferiori), e dal momento che si paralizzano gli arti inferiori non devono passare più di 24-48 ore per l'intervento. Bisogna intervenire entro 24-48 ore! Se passa più di questo tempo il paziente non recupera più l'uso degli arti inferiori. Questa è una cosa che il prof non capisce perché i neurochirurghi non sanno o non vogliono sapere, ma quando un paziente ha una compressione midollare e passano più di 48 ore senza aver risolto quella compressione midollare che provoca una paralisi, il paziente non recupera più l'uso delle gambe. Quindi questa è una emergenza! Il paziente che ha una compressione midollare tale che provoca la paralisi degli arti inferiori o paralisi degli arti superiori, dipende da dove interviene la compressione, quella è un'emergenza chirurgica, medica, radiologica, bisogna fare subito una risonanza, bisogna fare subito o una chemioterapia o una radioterapia o un intervento chirurgico, ma bisogna agire subito! (slide) Qui vedete altre localizzazioni scheletriche soprattutto appunto della colonna di pazienti con lesioni focali della colonna. Qui vedete un pattern iperintenso, tutte queste zone bianche non sono altro che localizzazione di malattia a livello della colonna. Qui vedete questa vertebra che ha perduto del tutto la sua morfologia, è ridotta a un cuneo rispetto ad una vertebra normale. Questo è un quadro drammatico di schiacciamento vertebrale con compressione del midollo spinale, è un quadro di un paziente che ha una paralisi completa degli arti inferiori. Domanda di una collega: “Qual è il segmento vertebrale che viene maggiormente compromesso?” Risposta: “Generalmente il tratto più basso della colonna dorsale, T12... però a volte anche L1-L2 sono spesso compromesse. Quindi livello basso, usualmente da T10 in giù. Però abbiamo avuto anche pazienti che hanno avuto schiacciamento di C2, quindi con compressione molto alta e quindi con una paralisi completa degli arti sia superiori che inferiori. Però nella stragrande maggioranza dei casi da T10 a L2, che sono le zona di maggior carico della colonna fondamentalmente per cui l'osteoporosi crea maggior facilità che queste vertebre si schiacciano e comprimano”. Qual è la patogenesi delle lesioni osteolitiche? Come detto nel caso del mieloma non c'è solo una attivazione osteoclastica e quindi di conseguenza la lesione osteolitica, ma c'è anche l'inibizione dell'attività osteoblastica per cui la lesione osteolitica si forma e non viene più riparata. Noi abbiamo anche pazienti che hanno la scomparsa completa della malattia mielomatosa, ma che rimangono con le lesioni osteolitiche perché queste lesioni osteolitiche non essendoci l'attività osteoblastica non vengono riparate. Tanti fattori possono indurre riassorbimento osseo nel mieloma, la IL-6 stessa che è il fattore di crescita delle plasmacellule che può indurre attivazione osteoclastica, ma ci sono una serie di altri fattori che è inutile elencare ma che fondamentalmente determinano una attivazione osteoclastica con liberazione appunto del calcio. C'è un modello importante che volevo sottolineare, il fatto che tutti questi fattori osteoclastogenici in pratica agiscono più spesso attraverso le cellule stromali, cioè non è una azione diretta sugli osteoclasti, ma è una azione mediata dalle cellule stromali. Le cellule stromali, a contatto con questi fattori osteoclastogenici che vi ho fatto vedere prima, IL-6. IL-11, MIP-1α esprimono sulla loro superficie una maggiore quantità di ligando RANK Il ligando RANK va a legarsi ai precursori degli osteoclasti determinando la loro proliferazione e la loro attivazione. Quindi questi osteoclasti che poi derivano dalle cellule monocitarie fondamentalmente, sono macrofagi, vengono attivati da RANK-ligand che lega appunto il recettore RANK che attiva poi tutta una serie di messaggi tipo NFkB ed altri che determina una proliferazione ed attività degli osteoclasti. Esiste in natura un cosiddetto “decoy receptor”, un recettore trappola che è la osteoprotegerina (OPG) che compete con i RANK presenti sugli osteoclasti per questo ligando, quindi quando c'è un equilibrio tra OPG e RANK c'è una corretta distribuzione e quindi una corretta attivazione di osteoclasti. Nel mieloma è stato dimostrato che c'è : − una riduzione di osteoprotegerina, quindi viene meno questo recettore trappola per RANK ligando; − una maggiore produzione di RANK ligando da parte dei fattori osteoclastogenici. Queste due sono le condizioni che poi attivano i precursori degli osteoclasti e quindi l'attività degli osteoclasti. Un altro fattore è il MIP-1α e quindi fondamentalmente ci sono diverse vie che poi attivano questi osteoclasti. Gli osteoclasti ad opera di alcune proteasi, le metalloproteinasi, le catepsine, ad opera di alcune pompe protoniche ecc.. possono poi determinare tutta una digestione della matrice ossea che è poi responsabile della lesione osteolitica. Lezione 11 Vi ricordo la distinzione che esiste tra la gammapatia monoclonale di incerto significato e il mieloma, fondamentalmente per fare diagnosi di mieloma multiplo deve esserci la compromissione d’organo oltre che un aumento della componente monoclonale e dell’infiltrato plasmacellulare. Nella lezione precedente è stata presa si è parlato della stadiazione secondo Durie e Salmon, della stadiazione secondo l’International Staging System ed è stata vista l’importanza della citogenetica, sia convenzionale che in FISH. E’ stato visto come in effetti i pazienti con citogenetica iperdiploide vanno meglio dei non iperdiploidi; abbiamo visto la sintomatologia di presentazione soprattutto con i dolori ossei: E’ stata analizzata poi qual è la patogenesi dei sintomi del mieloma multiplo: l’infiltrazione midollare, i sintomi legati alla componente monoclonale (in particolare l’iperviscosità), i sintomi legati al riassorbimento osseo (in particolare le lesioni osteolitiche e l’ipercalcemia), abbiamo visto come l’ipercalcemia è una sindrome che deve essere ricercata, deve essere attentamente valutata perché può dare dei sintomi molto importanti che a volte sono molto aspecifici e di difficile diagnosi → la sindrome ipercalcemica peraltro provoca un peggioramento della funzionalità renale ed è stato visto come l’insufficienza renale è una delle caratteristiche tipiche del mieloma multiplo. Ricordiamo inoltre il deposito delle catene leggere che provoca una condizione di tipo amiloidotico; la possibile sindrome neurologica sia per danno dei nervi periferici (quindi una polineuropatia periferica) sia per compressione delle radici nervose e soprattutto del midollo spinale. Abbiamo visto come nella diagnostica delle lesioni ossee siano importanti alcuni esami di imaging ma non la scintigrafia convenzionale. Per concludere è stata vista la patogenesi delle lesioni ossee dove c’è un’attivazione dei precursori osteoclastici da parte del RANK ligand per contemporanea riduzione dell’osteoprotegerina e per attivazione di altri fattori come il MIP-1α. Prognosi Quindi i pazienti che hanno delle lesioni ossee vanno sicuramente molto peggio rispetto a chi non le possiede, ma anche il pattern alla RMN può avere importanza prognostica, se è un pattern di tipo nodulare o diffuso o ambedue. Trattamento Come visto possiamo distinguere il mieloma cosiddetto smouldering o indolente dal mieloma multiplo e soltanto il mieloma multiplo è la condizione che deve essere trattata. Quindi le indicazioni al trattamento sono soprattutto: • Presenza dei sintomi • presenza di osteolisi • abbassamento dei valori di emoglobina • aumento della creatinina • concentrazioni elevate di β2-microglobulina • presenza di una grossa quantità di catene leggere nelle urine cioè la proteina di Bence-Jones • quando le plasmacellule midollari sono superiori al 30% • quando c’è una rapida progressione della malattia I vecchi farmaci che noi utilizziamo soprattutto: 1. agenti alchilanti 2. antraciclina 3. steroidi, sono molto efficaci nella terapia del mieloma multiplo, ma chiaramente da soli non sono in grado di dare una buona risposta. In effetti se noi consideriamo gli studi fatti (il prof illustra una vecchia pubblicazione del 1998), quando si utilizzava soltanto il vecchio Melphalan + Prednisone, chiamato con la sigla MP. Il melphalan è un agente alchilante, combinato con un cortisonico: questa combinazione è stata messa a confronto con numerosi studi che hanno valutato l’efficacia di regimi poli-chemioterapici attivi contro il mieloma, e se questi regimi poli-chemioterapici hanno dato una percentuale di risposta leggermente superiore rispetto al classico MP → in realtà se poi andiamo a vedere la curva di durata della risposta e la curva di mortalità tra i regimi di polichemioterapia e i regimi di melphalan e prednisone non c’è alcuna differenza, addirittura le due curve sono sovrapponibili. Il regime con MP è stato utilizzato sin dal 1968, questo significa che dal 1968 al ’98 non c’è stato nessun miglioramento in termini di efficacia nel far perdurare una risposta al trattamento e nel migliorare la sopravvivenza. Successivamente invece l’impiego di alte dosi di chemioterapie con il trapianto di midollo hanno permesso di ottenere dei risultati sicuramente migliori in termini di sopravvivenza (anche se non c’è un plateaux). Quindi sicuramente c’è un vantaggio di sopravvivenza indotta da alte dosi di chemioterapia che però si manifesta soltanto dopo 30 mesi di follow up ma che purtroppo come detto non è associato a un plateaux, vuol dire che se noi facciamo un trattamento ad alte dosi di chemioterapia (che al momento rappresenta il trattamento standard per i pazienti affetti da mieloma) guadagniamo qualcosa in termini di sopravvivenza ma non facciamo guarire questi pazienti. Il trapianto di midollo si può distinguere fondamentalmente in: 1. Autologo, vengono utilizzate le stesse cellule staminali midollari del paziente. 2. Allogenico, vengono trasfuse delle cellule staminali del midollo di un donatore. La differenza sta nel fatto che il trapianto autologo non è un vero e proprio trapianto, ma un modo per poter fare più chemioterapia → la chemioterapia infatti è tossica per il midollo, per le cellule staminali quindi noi prendiamo le cellule staminali del paziente (le mettiamo da parte), poi facciamo un mega-chemioterapia al paziente → questa distrugge tutte il midollo del paziente, tranne le cellule midollari staminali che erano state messe da parte. Quindi trapiantiamo queste cellule, e queste ripopolano il midollo perché non avevano subito l’insulto della chemioteapia. Questo è grosso modo il concetto dell’autotrapianto. Viceversa nel trapianto allogenico immettiamo cellule staminali di un donatore che deve essere HLA identico. Quindi nel primo caso è una sorta di stratagemma per poter fare più chemioterapia, nel secondo caso invece c’è un effetto immunologico in quanto le cellule midollari del donatore sono cellule immuno-competenti quindi trasfondiamo insieme al midollo tessuto immuno-competente che quindi agisce in maniera immunologica nei confronti della malattia del ricevente, ha un effetto di aggressione immunologica nei confronti della malattia del paziente. Quindi nel primo caso del trapianto autologo abbiamo soltanto l’effetto delle alte dosi di chemioterapia per combattere la malattia, invece nel caso del trapianto allogenico oltre alle alte dosi di chemioterapia abbiamo anche un effetto immunologico, un’aggressione immunologica da parte del tessuto immuno-competente del donatore sano nei confronti della malattia. Questa aggressione immunologica però non è del tutto specifica nei confronti della malattia neoplastica ma è anche un’aggressione immunologica nei confronti di tutto l’organismo ricevente → infatti questa condizione si chiama graft versus host (GVH), cioè una sorta di “rigetto al contrario” , quindi non è il ricevente che rigetta l’organo trapiantato ma è l’organo trapiantato che aggredisce il ricevente. Pertanto questo può provocare tutta una serie di condizioni morbose che possono portare anche alla morte del paziente. Ciò fa si che in realtà il trapianto allogenico (in particolare nel mieloma multiplo) è ancora gravato da un’elevata mortalità, quindi se il trapianto allogenico è sicuramente un trattamento efficace nei confronti del MM, tuttavia è limitato ai pazienti particolarmente giovani. Intorno al 20% dei pazienti che fanno il trapianto allogenico per MM muoiono per la procedura trapiantologica. Quindi in definitiva le curve di autotrapianto e allotrapianto si equivalgono, nel senso che nell’autotrapianto vi sono molte più ricadute rispetto all’allotrapianto, tuttavia nell’allotrapianto ci sono molte più morti per procedura. Anche se nel tempo ci sono stati dei miglioramenti, tuttavia questi non sono sufficienti a consigliare un trapianto allogenico a tutti i pazienti affetti da mieloma, il trapianto allogenico al momento è confinato solo a pazienti: • Giovani • E a determinati studi clinici Quello che invece è cambiato nello scenario del trattamento del Mieloma Multiplo è stato l’avvento di nuovi farmaci che agiscono su uno dei meccanismi patogenetici più importanti nel mieloma multiplo → nel MM le plasmacellule crescono non soltanto per proliferazione spontanea neoplastica ma soprattutto perché nel midollo interagiscono con le cellule stromali e qui intervengono dei meccanismi che fanno proliferare e crescere le plasmacellule: • questi nuovi farmaci interferiscono proprio in quelli che sono i meccanismi di legame tra le plasmacellule e le cellule stromali midollari. • inoltre questi nuovi farmaci agiscono su alcune caspasi, cioè in diversi punti delle vie dell’apoptosi e quindi possono avere un’azione sinergica tra di loro. Uno dei farmaci più importanti per il trattamento del Mieloma Multiplo è il Bortezomib che è un inibitore del proteosoma. E’ stato dimostrato che le plasmacellule crescono anche per via di un fattore molto importante che è NF-kB, questo è un vero e proprio motore della cellula neoplastica perché è in grado di stimolare la progressione del ciclo cellulare, è in grado di produrre citochine e perché è in grado di stimolare una serie di cascate che hanno un effetto anti-apoptotico. Tutto questo l’NF-kB lo fa quando entra nel nucleo: in condizioni normali però l’NFkB sta nel citoplasma e qui è legato all’inibitore IkB, il quale legandosi all’NFkB impedisce l’entrata di questo nel nucleo. Per effetto di alcune condizioni, di alcuni stimoli, l’IkB viene degradato dal proteosoma e così facendo libera l’NFkB che può entrare nel nucleo ed esercitare la sua funzione di stimolazione della crescita neoplastica. Il Bortezomib blocca il proteosoma e quindi la degradazione dell’IkB → così facendo mantiene il legame con l’NFkB e non permette l’entrata dell’NFkB nel nucleo. Il Bortezomib appartiene quindi a una classe di farmaci che sono gli inibitori del proteosoma. Il proteosoma come sappiamo è una sorta di frullatore delle proteine, serve per la digestione delle proteine, in questo caso è l’IkB. È un farmaco attivo nei confronti del mieloma. Bisogna tenere presente che circa il 70% dei pazienti trattati con questo farmaco ha avuto una risposta positiva al trattamento, considerando che si tratta con pazienti refrattari o ricaduti alle terapie convenzionali. Come agisce il cortisone? * Il cortisone invece agisce soprattutto sulle caspasi, il desametasone agisce sulla caspasi 9 e attiva i meccanismi di apoptosi. Il cortisone è uno dei più efficaci agenti citolitici nei confronti delle plasmacellule neoplastiche, però non riesce ad agire su questo sistema, cioè il cortisone agisce prevalentemente sulle plasmacellule dove ne determina morte per apoptosi, tuttavia non riesce a interferire con i meccanismi di interazione tra plasmacellule e cellule stromali, cosa che invece riescono a fare con altri meccanismi gli inibitori del proteosoma (Bortezomib) → e soprattutto ci riescono i cosiddetti “Imids” cioè la Talidomide e la Lenalidomide. La talidomide, il famoso farmaco teratogeno che negli anni ’60 ha provocato la focomelia, è stato rivalutato per il trattamento del Mieloma Multiplo appunto perché è in grado di interferire con il legame plasmacellule- cellule stromali. • oltre ad avere un effetto anti-angiogenetico (inibisce il VEGF); • effetto di immunomodulazione perché aumenta la produzione da parte dei linfociti di IFN-gamma e IL-2 • agisce sulle cellule stromali. La talidomide proprio per questo effetto di inibizione della crescita degli arti è stato testato negli anni ’60 nelle neoplasie in quanto farmaco che inibiva la crescita, però non è un farmaco che ha un grosso effetto anti-neoplastico in generale. In questi studi degli anni ’60 vi fu qualche paziente affetto da mieloma in cui vi era stato un arresto della proliferazione della malattia, ma questo evento era passato un po’ sotto silenzio: una decina di anni fa vi fu negli Stati Uniti un paziente con mieloma multiplo resistente al trattamento; la moglie di questo paziente si mise così su internet per cercare qualunque farmaco potesse trovare che aveva effetti sul mieloma e scoprì proprio questi studi vecchi degli anni ’60 che riportavano il caso di questo paziente affetto da mieloma in cui c’era stato un rallentamento della progressione della malattia trattata con talidomide. Dal momento che la talidomide viene utilizzata negli Stati Uniti per il trattamento dei noduli della lebbra era disponibile in commercio, questa donna convinse quindi il medico statunitense a provare la talidomide nel marito affetto da mieloma: questo paziente ha risposto al trattamento, anche se in maniera transitoria e da lì la talidomide è stata utilizzata in altri pazienti, in particolare si è visto che circa il 30% dei pazienti con MM trattati con talidomide rispondono al trattamento. Questo per capire come la talidomide è un farmaco la cui riscoperta è stata del tutto occasionale. Poi sono cominciati a proliferare vari studi in cui si dimostrava che la talidomide come agente singolo era in grado di dare una risposta positiva nel 30-40% dei pazienti ricaduti o refrattari e poi da lì tutta una serie di combinazioni di taloidomide e desametasone oppure talidomide con altri farmaci in combinazione. La talidomide è quindi sicuramente un farmaco efficace anche se non è privo di effetti collaterali, in particolare gli effetti collaterali della talidomide sono legati al tromboembolismo, la tromboflebite. La combinazione talidomide + melphalan-prednisone è molto più efficace del solo melphalan-prednisone tradizionale, però con questa problematica delle tromboflebiti: tant’è che adesso se si usa la talidomide bisogna fare una profilassi anti-trombotica o con l’eparina o cono la semplice aspririna. La Lenalidomide, derivato della talidomide, è un farmaco anche più efficace della talidomide in termini di citotossicità delle cellule mielomatose. • dà una mielotossicità di grado moderato quindi trombocitopenia e neutropenia; • forte attività anti-angiogenetica. • la lenalidomide è in grado anche di far aumentare le cellule NK, quindi effetto immunologico, aumentano il numero e l’attività delle cellule NK. Questo ha fatto sì che la combinazione Lenalidomide + Desametasone sia una delle combinazioni più attive sui pazienti affetti da mieloma multiplo. La Lenalidomide non provoca neuropatia rispetto alla sola Talidomide che invece la provoca. In studi sulla combinazione Lenalidomide + Melphalan-Prednisone la percentuale di pazienti che risponde già al primo ciclo di trattamento con questa combinazione è molto alta nei pazienti con mieloma: il 24% dei pazienti ottiene addirittura una risposta completa dopo il primo ciclo considerate invece che con le terapie tradizionali eravamo abituati a vedere dopo 6 cicli di trattamento una percentuale di remissione completa inferiore al 5%. Si capisce quindi come ci sono significative novità in termini di terapia per quanto riguarda il mieloma multiplo, tant’è che oggi stiamo testando la possibilità di non fare l’autotrapianto, neppure nei soggetti giovani, ma di trattarli con questi nuovi farmaci (in particolare Lenalidomide e inibitori del proteosoma) che danno percentuali di risposta assolutamente sovrapponibili a quelli dell’autotrapianto. Se combiniamo quindi la Lenalidomide + Bortezomib abbiamo i migliori risultati, tanto buoni che azzerano anche alcuni fattori prognostici negativi, ad es nei pazienti trattati con Bortezomib non vi è più l’importanza prognostica della delezione del 13, cosa che invece è presente nei pazienti trattati con terapia convenzionale (i pazienti trattati con terapia convenzionale che non hanno la delezione del 13 vanno meglio rispetto a quelli che hanno la delezione del 13). Quindi sono farmaci con meccanismi d’azione differenti, che possono essere utilizzati in combinazione e che superano l’impatto prognostico negativo di alcune alterazioni cromosomiche (quali la delezione del 13 o la 4;14), che possono essere utilizzati in pazienti con insufficienza renale e anche in pazienti anziani. Ma ancora abbiamo bisogno di migliorare di molto l’impiego di questi farmaci. Un ultimo aspetto del mieloma sono i farmaci che utilizziamo per la malattia ossea: Quali sono le opzioni terapeutiche nei pazienti che hanno malattia ossea? 1. Radioterapia 2. Chirurgia 3. Farmaci bifosfonati, sono quelli utilizzati con più frequenza, sono farmaci che hanno avuto un significativo miglioramento in termini di capacità terapeutica (dal vecchio etidronato fino all’acido zoledronico che ha una potenza 10000 volte superiore rispetto all’etidronato o al clodronato). Questi farmaci sono dunque quelli che utilizziamo nei pazienti con malattia ossea e hanno un meccanismo di azione peculiare in quanto agiscono soprattutto inibendo la formazione degli osteoclasti e la loro efficacia è stata dimostrata nei confronti del placebo, sia in pazienti con malattia ossea da mieloma, sia in pazienti con malattia ossea da metastasi di carcinoma della mammella. Questi farmaci hanno così migliorato la qualità della vita dei pazienti affetti da lesioni osteolitiche che probabilmente ne migliorano anche la quantità di vita, perché se un paziente non è più allettato, non si fa più la tromboflebite, riesce a vivere meglio probabilmente vive anche più a lungo! Accanto a questo è stato dimostrato per i bifosfonati un effetto anti-neoplastico in vitro, anche se l’effetto in vivo non è così evidente. L’unico problema dei bifosfonati sono gli effetti collaterali: la tossicità renale piuttosto modesta ma presenta, tuttavia l’effetto collaterale più importante osservato negli ultimi anni è stata la cosiddetta osteo-necrosi della mandibola. Purtroppo negli ultimi anni c’è stata una notevole incidenza di aumento di osteonecrosi della mandibola, ovvero una condizione di osteomielite dell’osso mandibolare che provoca l’esposizione dell’osso (non è più ricoperto da mucosa). Questo è correlato all’esposizione ai bifosfonati, ma è molto correlato anche all’igiene dentale e all’estrazione dei denti. Quindi è estremamente importante nei pazienti che fanno terapia con bifosfonati avere una buona igiene dentale e limitare le procedure di estrazione dentaria ai casi in cui non se ne può fare a meno. 4. Ci sono anche delle terapie sperimentali fatte su animali di laboratorio. Trattamento con osteo-protegenina (che ricordiamo è ridotta nel mieloma) ed è quella che blocca in qualche modo il RANK-ligando. L’osteoprotegenina permette quindi la ricostituzione dell’osso. 5. Un’altra procedura che viene fatta ormai routinariamente nei pazienti affetti da mieloma è la cosiddetta vertebro-plastica, questa si utilizza nei pazienti con schiacciamento vertebrale (si fa anche nell’osteoporosi). Nella vertebro-plastica si inietta del cemento, nella cifoplastica si fa una procedura ulteriore per cui si mette una sorta di ago dentro la vertebra, si gonfia un palloncino e dentro questo palloncino si inietta il cemento. Queste procedure ci permettono di stabilizzare delle vertebre schiacciate, le quali rappresentano una condizione molto frequente nei pazienti affetti da mieloma. Tutto questo ha permesso negli ultimi anni di ottenere un significativo miglioramento della sopravvivenza, però limitato ai pazienti di età < a 60 anni; questo è legato sì alla possibilità di fare il trapianto ma anche alla possibilità di utilizzare questi nuovi farmaci. I nuovi farmaci come la Talidomide, la Lenalidomide e il Bortezomib riescono quindi ad agire sulla storia naturale della malattia, anche se bisogna ricordare che ancora il mieloma è una malattia che non guarisce infatti come si vede nella curva di sopravvivenza non c’è un plateaux, quindi c’è un significativo guadagno rispetto ai pazienti trattati con vecchi farmaci, pazienti anche sottoposti ad autotrapianto, ma ancora non siamo in grado di far guarire questi pazienti. AMILOIDOSI primitiva Abbiamo accennato infatti all’amiloidosi secondaria, dove il processo dipende dal fatto che c’è il mieloma, vengono prodotte delle catene leggere che tendono a precipitare e danno queste compromissioni. Esiste anche una patologia che prende il nome di amiloidosi primitiva, una condizione che può essere dovuta a diverse patologie, in particolare ci occupiamo della cosiddetta Amiloidosi AL, - infatti vi possono essere anche delle altre condizioni di amiloidosi dove altre proteine anomale, che possono essere congenite o associate ad altre patologie, tendono a precipitare e quindi a dare insufficienza d’organo. Nell’amiloidosi AL l’insufficienza d’organo interviene a carico dei reni, cuore e fegato. • Sezione di cuore, con setto interventricolare ispessito e questo è dovuto alla deposizione di sostanza amiloide. Sono pazienti con deposito di amiloide: • A livello dell’intestino e quindi diarrea cronica • • • A livello dei nervi periferici e in particolare a livello del tunnel carpale (in pazienti con tunnel carpale in entrambe le mani sospettare sempre l’amiloidosi!!) Nella lingua determina una macroglossia da amiloide con impronta dei denti Lesioni purpuriche periorbitali che sono patognomoniche dell’amiloidosi, dipende dal fatto che i depositi amiloidi infarciscono la parete dei piccoli vasi e quindi questi pazienti hanno una particolare fragilità capillare e sono molto colpiti i vasi periobitali. L’amiloidosi AL è caratterizzata da una piccola componente monoclonale, in definitiva l’amiloidosi non è altro che un piccolo mieloma che però determina la produzione di catene leggere, le quali hanno una particolare tendenza alla precipitazione. Il rischio è che la componente monoclonale è così piccola che sfugge alla nostra osservazione oppure ci sembra una condizione non patologica. Ma se il paziente ha cardiomiopatia, insufficienza renale, diarrea, tunnel carpale bilaterale e macroglossia, fragilità vascolare pensiamo alla possibilità che il paziente possa avere l’amiloidosi. LEUCEMIE ACUTE Le leucemie acute sono un capitolo molto importante dell’ematologia perché costituiscono la patologia ematologica forse più impegnativa per diversi motivi. Le leucemie acute sono delle neoplasie maligne di trasformazione delle cellule ematopoietiche staminali che portano alla proliferazione di cellule blastiche. Distinguiamo le leucemie: 1. ACUTE 2. CRONICHE Entrambe suddivise in mieloide e linfoide. * Già abbiamo visto per quanto riguarda le leucemie croniche la leucemia linfatica cronica e in seguito parleremo della leucemia mieloide cronica Le leucemie acute possono essere quindi mieloide o linfoide in base allo stipite cellulare interessato e sono caratterizzate da una rapida proliferazione e dalla presenza di cellule immature. Ricordiamo che la ematopoiesi è caratterizzata dalla presenza di cellule pluripotenti, cellule staminali che poi si differenziano in mieloidi o linfoidi e a seconda di dove cade la lesione che induce la leucemia, questa può essere linfoide o mieloide. Epidemiologia La Leucemia acuta linfoide (LAL) è tipica del bambino, l’incidenza è quasi esclusivamente limitata all’età giovane sino ai 14 anni, invece è molto bassa nell’adulto per poi avere un lieve aumento nei soggetti con età superiore ai 65 anni. La Leucemia Acuta Mieloide viceversa è molto rara nel bambino ma è molto frequente nell’anziano, quindi sono due malattie totalmente differenti in termini di incidenza. Leucemia acuta linfoide → tipica del bambino Leucemia acuta mieloide → tipica dell’anziano Fattori predisponenti: Sicuramente vi sono delle condizioni che predispongono allo sviluppo di queste malattie: • radiazioni ionizzanti • benzene, farmaco tossico per il midollo • alcune condizioni genetiche predisponenti (sindrome di down) In condizioni normali abbiamo le cellule staminali che si differenziano e poi danno luogo alla linea eritroide, alla linea granulocitica e monocitica e alla linea megacariocitica (anche se in questo caso trattiamo la leucemia acuta mieloide il discorso è lo stesso anche per quella linfoide). Quando c’è una trasformazione leucemica di una cellula staminale questa da un lato prolifera e dà luogo a un aumento dei leucociti ed una infiltrazione d’organo e quindi compromissione d’organo; dall’altro non può differenziarsi in normali cellule e inibisce il differenziamento delle normali cellule: quindi il blasto leucemico da un lato prolifera ma dall’altro inibisce la normale mielopoiesi. Quello che osserviamo in questi pazienti è una iperleucocitosi molto spesso costituita soltanto da cellule blastiche con compromissione d’organo e dall’altro lato l’inibizione della normale emopoiesi e quindi la riduzione delle piastrine, dei globuli bianchi e rossi. Tutto questo porta a quelli che sono i sintomi fondamentali della leucemia: • infezioni • emorragie • compromissione d’organo, che può portare a morte Quindi la patogenesi della leucemia è una condizione in cui c’è questo clone neoplastico che risponde poco ai normali meccanismi regolatori, essendo un clone neoplastico, e che riduce la capacità del normale differenziamento, accumulandosi sia nel midollo che nel sangue periferico e sopprimendo la normale mielopoiesi. Sintomatologia I sintomi sono legati alla sostituzione delle cellule normali e quindi: • insufficienza midollare e quindi anemia- neutropenia- piastrinopenia • infiltrazione dei parenchimi, quindi organo-megalia e deficit funzionali • liberazione di sostanze attive da parte dei blasti-leucemici e quindi: febbre e CID che è una grave coagulopatia che può accompagnare le leucemie acute. Quindi è presente: 1. iperproduzione di blasti, generalmente superiore al 30% nel midollo 2. ridotta presenza di cellule normali: GR e quindi anemia- GB neutropenia- ridotta trombocitopoiesi e quindi piastrinopenia 3. Possono essere interesssati anche i siti extra-midollari, quali linfonodi-milza-fegato..etc Come facciamo la Diagnosi di Leucemia Acuta? 1. La diagnosi la facciamo attraverso la valutazione citologica del sangue periferico e del midollo 2. Attraverso l’immunoistochimica - nel caso di leucemia acuta mieloide andando a marcare queste cellule con alcuni anticorpi per la mieloperossidasi, vediamo che sono positive per queste mieloperossidasi oppure presentano esterasi non specifiche che sono indicative di una origine monocitica. 3. Ma soprattutto la diagnosi la facciamo attraverso la valutazione in citofluorimetria degli antigeni che marchiamo con gli anticorpi, abbiamo: • antigeni molto precoci come il CD34 • antigeni appartenenti alla linea mieloide CD13 e 33 • oppure nelle leucemie megacariocitiche la positività per il CD41 e 61 4. citogenetica 5. valutare la biologia molecolare di queste cellule Anatomia patologica Quadro morfologico di leucemia acuta: le cellule sono grossomodo abbastanza monomorfe, cioè sono tutte uguali. Questo è una provetta con sangue, in caso di LA particolarmente ipercitosica il plasma è molto più chiaro (a indicare una chiara alterazione del plasma), ma soprattutto ci sono pochissimi GR e molti GB che giustificano il termine di leucemia. Leucemia significa infatti “sangue bianco”. Classificazioni: Per le Leucemie Acute mieloide e linfoide vi sono diverse classificazioni: 1. la prima è la cosiddetta classificazione FAB (french- american- british) che distingue le LA mieloide a seconda delle caratteristiche morfologiche in 8 sottotipi, da M0 a M7. Le prime 4 appartengono alla linea granulocitaria, 3 appartengono alla linea monocitaria, la M6 alla linea eritroide e la M7 alla linea megacariocitica. • M0 non c’è nessuna differenziazione granulocitaria • M1 modesta differenziazione granulocitaria (granuli nel citoplasma indica un certo differenziamento) • M2 buona differenziazione granulocita ria (comparsa di corpi di Auer) • M3 sono tutti bloccati a livello di pro- mielocita che è uno stadio abbastanza avanzato dell’evoluzione lungo la linea granulocitica (fasci di corpi di Auer) • M4 Mielo-monocitica ( cellule con morfologia di tipo granulocitario e anche monocitario) • M5a monocitica-immatura o monoblastica (solo monociti) M5b monocitica matura • M6 eritroleucemia, l’alterazione leucemica interessa la linea eritroide • M7 l’alterazione leucemica interessa la linea megacariocitica Vi possono essere delle localizzazioni extra-midollari di malattia: • Gengive, soprattutto nelle leucemie acute della linea monolitica, pertanto quando vediamo un paziente con ipertrofia gengivale e con alterazioni dell’emocromo prendiamo in considerazione di mandarlo dall’ematologo • Localizzazioni cutanee di malattia Alterazioni citogenetiche La valutazione citogenetica è importante perché esistono delle alterazioni citogenetiche: favorevoliintermedie – sfavorevoli. 1. Alterazioni citogegenetiche favorevoli sono: traslocazione t(15;17); la inversione del 16; la traslocazione t(8;21) 2. Intermedie: sono rappresentate dal cariotipo normale 3. Sfavorevoli: delezione del 5, del 7 e il cariotipo complesso. Questo è importante per capire quanto dobbiamo aggredire la malattia: • perché se il paziente ha delle alterazioni citogenetiche che gli configurano un cariotipo intermedio o sfavorevole, è un paziente che deve andare in conto a trapianto di midollo (se non è anziano). • Viceversa i pazienti che hanno un cariotipo favorevole, possono non subire il trapianto di midollo allogenico (una procedura complessa e anche gravata da una certa mortalità) Vengono mostrate le curve di sopravvivenza di pazienti con leucemia mieloide acuta trattati con la chemioterapia e trapianto di midollo: si comportano in maniera diversa, in base alla citogenetica, si possono osservare grandi differenze. Quindi la stessa patologia dal punto di vista morfologico, ma alla cui base si riscontrano delle anomalie citogenetiche differenti, si comporta in maniera del tutto differente. Inoltre dobbiamo considerare che i pazienti di età più avanzata sono quelli che hanno le alterazioni citogenetiche più sfavorevoli. Quindi l’ipotesi che si sente spesso dire in giro, cioè che la leucemia acuta dell’anziano è una malattia meno grave perché cresce più lentamente…è assolutamente una favola! anzi la Leucemia Acuta dell’anziano è proprio quella che non risponde al trattamento perché si associa alle peggiori alterazioni citogenetiche. In definitiva il paziente anziano non solo non può fare una terapia aggressiva ma presenta anche una malattia peggiore, questo fa sì che se da un lato abbiamo degli ottimi risultati per quanto riguarda i pazienti giovani affetti da leucemia acuta, dall’altro lato il trattamento della LA nell’anziano è una tragedia perché non spostiamo di nulla la curva di sopravvivenza. Quindi è molto importante fare in questi pazienti la valutazione genetica e molecolare perché ci permette di fare un corretto inquadramento diagnostico e riconosce di età biologiche e cliniche differenti, anche se sono tutte quante all’interno di una stessa classificazione FAB. Dunque non ha più importanza se il paziente ha una classificazione FAB M1 o M2..etc ma l’aspetto importante è il tipo di citogenetica, è un fattore estremamente importante in termini prognostici e quindi viene utilizzato per una corretta stadiazione dei pazienti. Il prof mostra le curve di sopravvivenza dei pazienti anziani, dove non c’è stato alcun miglioramento negli ultimi anni; la differenza con le curve di sopravvivenza nei pazienti giovani dipende fondamentalmente dalla citogenetica e dalla possibilità che abbiamo di trattare i pazienti con terapie abbastanza aggressivi, tuttavia bisogna notare che anche pazienti giovani con citogenetica sfavorevole hanno curve di sopravvivenza uguali a quelle dell’anziano. Lezione 12 LEUCEMIA ACUTA MIELOIDE – Riassunto lezione precedente La volta scorsa abbiamo iniziato a parlare della leucemia acuta, in particolare della leucemia mieloide acuta e vi ho fatto vedere qual è la patogenesi dei sintomi nelle leucemie acute: i sintomi sono legati al fatto che questi blasti, le cellule leucemiche, da un lato proliferano e circolano nel sangue e negli organi coinvolti, dall'altro lato non si possono differenziare e inibiscono la differenziazione delle cellule normali e quindi alla fine c'è anemia, granulocitopenia e piastrinopenia. Si è visto qual è la classificazione morfologica delle leucemia acute mieloidi e soprattutto l'importanza della citogenetica: ci sono delle lesioni citogenetiche che determinano una prognosi favorevole, delle lesioni che determinano una prognosi intermedia e infine delle lesioni citogenetiche che determinano una prognosi fortemente negativa. E’ stato visto come pure le lesioni che sono le più negative come la delezione del 5 o la delezione del 7, sono appannaggio dell'età più anziana, mentre le lesioni citogenetiche più favorevoli, come l'inversione del 16, la t(15;17), la t(8;21) sono appannaggio dell'età più giovane. Quindi bisogna sottolineare il fatto che le alterazioni cromosomiche nelle leucemie acute mieloidi costituiscono l'elemento diagnostico e prognostico più importante perché queste alterazioni cromosomiche e molecolari servono per la diagnosi, perché permettono un corretto inquadramento diagnostico e perché all'interno della classificazione FAB riescono a identificare delle entità biologiche e cliniche ben distinte, e sono poi molto importanti perché costituiscono un fattore prognostico indipendente, anzi è il fattore prognostico importante per le leucemie acute. Quindi abbiamo soprattutto: - delle traslocazioni, che costituiscono il 40%, - il 10% sono anomalie numeriche, - e poi le delezioni sono il 4%. Ci sono tutta una serie di mutazioni di geni, per es il FLT3, il Ras, il c-kit etc che sono variamente rappresentati ma quello più importante è la mutazione del FLT3. Queste alterazioni citogenetiche possono essere visualizzate in citogenetica convenzionale, ma possono essere visualizzate anche con FISH e in biologia molecolare. Le delezioni cromosomiche sono molto più frequenti nell'anziano e fondamentalmente determinano la perdita di un gene oncosoppressore, per cui questa delezione fa perdere un gene oncosoppressore e frequentemente esiste anche una mutazione sul cromosoma omologo che è apparentemente normale e quindi quel gene è del tutto mancante, poiché manca un pezzo di cromosoma in un allele e perché l'altro allele ha spesso delle mutazioni. Però non sempre noi riusciamo a individuare delle alterazioni cromosomiche nei pazienti con leucemia acuta: ci sono molti pazienti nei quali la citogenetica ci dà un cariotipo normale, questo non vuol dire che non ci sono alterazioni del DNA, vuol dire semplicemente che la citogenetica convenzionale non riesce ad evidenziare alcune delezioni, alcune piccole traslocazioni chee chiaramente hanno bisogno di metodiche più sofisticate. Però esiste un gruppo di leucemie acute mieloidi a cariotipo normale e quindi per questi pazienti occorre cercare altri parametri prognostici. Questo è un gruppo abbastanza eterogeneo: 1) in alcuni si riscontra l’alterazione del gene FLT3, in particolare abbiamo la cosiddetta Internal Tandem Duplication, vuol dire una duplicazione interna di questo che in pratica determina un'attività proliferativa della cellula. Il FLT3 è in pratica un recettore per i fattori di crescita: per essere attivo ci vuole una dimerizzazione di due molecole del FLT3, succede quindi che per una duplicazione interna c'è una attivazione costitutiva del FLT3, per cui è come se questa cellula subisse sempre l'effetto di un fattore di crescita; - un'altra possibile mutazione (si tratta di mutazioni puntiformi), ha fondamentalmente lo stesso meccanismo, cioè sempre di attivazione, di dimerizzazione e quindi di attivazione del recettore. Quindi le modifiche di questo gene fanno si che questa cellula assuma un vantaggio proliferativo perché è come se il recettore per il fattore di crescita fosse sempre stimolato. Questo è un aspetto presente in molti pazienti che hanno una leucemia acuta mieloide a cariotipo normale: in questi pazienti bisogna andare a ricercare se c'è l'alterazione del FLT3, e questa alterazione configura un rischio elevato per cui questi pazienti devono essere sottoposti a terapie molto aggressive perché l'autotrapianto non è in grado di far guarire questi pazienti. Il FLT3 è appunto associato ad un cariotipo normale. 2) L’altra alterazione che è frequentemente associata a cariotipo normale nelle leucemie acute è la alterazione della nucleofosmina. Il gene della nucleofosmina (NPM), contrariamente al FLT3 configura invece una prognosi favorevole. Per cui fondamentalmente quando abbiamo pazienti con leucemia acuta mieloide e cariotipo normale, dobbiamo andare a studiare in questi pazienti l’NPM1 e il FLT3 e distinguiamo questi pazienti in quattro tipi: NPM1+ FLT3+ NPM1+ FLT3NPM1- FLT3+ NPM1- FTL3-. Questo è molto importante perché in molti pazienti, 35% circa dei pazienti che hanno un leucemia acuta mieloide a cariotipo normale hanno questa alterazione dell’NPM1, frequentemente associato a cariotipo normale. Questi pazienti hanno un profilo abbastanza tipico: sono usualmente donne, pazienti con alta conta di globuli bianchi, hanno una diagnosi che è più spesso nella classificazione FAB M1, oppure M4 o M5 e una caratteristica importante è che sono CD34 negative, il CD34 come sappiamo è l’antigene che configura le cellule staminali, quindi è possibile che questa leucemia acuta insorga in cellule che sono più avanti nel programma maturativo della linea mieloide. La nucleofosmina è una proteina che si trova usualmente nel nucleo, quando è alterata passa nel citoplasma e questo può essere evidenziato, non c’è nemmeno bisogno di fare un’indagine di biologia molecolare per vedere se questi pazienti hanno la nucleofosmina alterata, basta andare a fare una colorazione morfologica e si vede come se la nucleofosmina si trova nel citoplasma, allora quella è una nucleofosmina mutata e che quindi c’è l’alterazione del gene della nucleofosmina. (Questa è una scoperta fatta da un italiano a Perugia che si chiama Michelangelo Farini, col quale l’università di Catania collabora molto nella diagnostica delle leucemia acute). Quindi l’altra alterazione, dicevamo, è il FLT3 e questo è importante perché questo FLT3 ha la capacità di trasformare, di determinare un’attivazione della crescita cellulare, il FLT3 ha un’importanza prognostica negativa: pazienti con la internal tandem duplication positiva vanno peggio rispetto e chi non ha la internal tandem duplication positiva e allora possiamo differenziare questi pazienti sulla base della NPM1 e sulla base del FLT3, perché abbiamo visto che questi sono pazienti che hanno un cariotipo normale: quindi noi abbiamo detto tutte le alterazioni citogenetiche sfavorevoli, tutte le alterazioni citogenetiche favorevoli ci permettono di fare una valutazione prognostica, ma quando il paziente ha un cariotipo normale dobbiamo usare questi parametri che sono la nucleofosmina e il FLT3 perché questi ci danno la prognosi a questo punto; quando abbiamo pzt che hanno una mutazione per la nucleofosmina che ha un valore prognostico favorevole, questa è la curva di sopravvivenza dei pazienti (immagine); viceversa i pazienti che sono FLT3 positivi hanno una prognosi nettamente sfavorevole; prognosi intermedia ce l’hanno i pazienti che sono negativi per il FLT3 e negativi per la NPM, perché la positività l’NPM ha una prognosi favorevole, la positività del FLT3 ha una prognosi sfavorevole: chi non ha nessuna positività né per l’NPM né per il FLT3 ha una prognosi intermedia e questo è importante perché sulla base di questo dobbiamo decidere la terapia, per cui pzt che sono + per la NPM e - per la FLT3 non li mandiamo al trapianto di midollo osseo, viceversa questi i pazienti NPM- e FLT3+ vengono mandati al trapianto di midollo osseo. 3) Un’altra alterazione può essere quella a carico di un gene che si chiama MLL, che si trova sul cromosoma 11 ed è importane perché più spesso questo gene è coinvolto in quelle leucemie acute cosiddette secondarie, esistono delle leucemie acute che sono secondarie, non insorgono de novo, improvvisamente ma insorgono in pazienti che hanno avuto un precedente disordine ematologico, oppure in pazienti trattati per altre neoplasie, per cui pazienti trattati con antiblastici per altre patologie oppure con radioterapia per altre neoplasie, hanno una maggiore incidenza di sviluppare delle leucemie, in quanto sia i farmaci antiblastici sia la radioterapia hanno un effetto mutageno e quindi possono favorire l’insorgenza di seconde neoplasie, in particolare possono favorire l’insorgenza di leucemie. Queste leucemie secondarie, che hanno una prognosi nettamente peggiore rispetto a quelle de novo, sono caratterizzate molto spesso da alterazioni del cromosoma 11 in particolare di questo gene MLL, che poi è un gene promiscuo perché nel suo riarrangiamento si riarrangia con 80 partner diversi,, cioè ci sono tutta una serie di possibili partner in queste traslocazioni che vanno a finire vicino al gene MLL e la variabilità è ben dimostrabile. Classificazione Leucemie Acute Vedendo un po’ la classificazione FAB della lezione precedente, a questa classificazione, che ricordiamo va: - M0 dalla leucemia mieloide senza differenziazione, - M1 con minima differenziazione, - M2 con elevata differenziazione - M3 promielocitica, - M4 e poi quelli della linea mielo-monocitica, - la M5, la monocitica pura, - la M6, la eritroide pura e - la M7, la megacariocitica pura, a questa definizione morfologica bisogna aggiungere le definizioni che ci vengono dalla citogenetica e che spesso combaciano con quella morfologica, per esempio la M2 spesso ha la traslocazione t(8,21), invece la M4 con eosinofili spesso ha questa alterazione citogenetica che è l’inversione del 16 e queste alterazioni citogenetiche correlano con il coinvolgimento di alcuni geni che sono poi importanti nella genesi di questa trasformazione neoplastica. Per cui adesso sono state fatte delle nuove classificazioni per le leucemie acute che non sono soltanto basate sulla morfologia, ma sono basate sulla combinazione della morfologia più la citogenetica e la biologia molecolare. Ora senza andare nei dettagli di queste classificazioni che sono molto specialistiche (queste sono classificazioni che cambiano sempre, ne è stata fatta una nuova nel 2008), fondamentalmente il concetto è che non è più un fatto soltanto morfologico ma è una classificazione che comprende sia la morfologia che anche le alterazioni citogenetiche. In questa classificazione ad es. viene considerata la leucemia acuta mieloide con queste anomalie genetiche ricorrenti, poi c'è la leucemia acuta mieloide con una displasia multilineare, cioè dove la malattia non solo determina una proliferazione di blasti mieloidi, ma c'è una alterazione morfologica delle normali serie: la serie eritroide, la serie megacariocitica e il residuo di cellule mieloidi che hanno delle alterazioni cosiddette displastiche, cioè delle alterazioni della maturazione. Poi c’è la leucemia acuta mieloide correlata alla terapia, cioè una precedente chemioterapia per un’altra malattia neoplastica può favorire l’insorgenza di neoplasie e in particolare questa categoria di leucemia acuta mieloide viene ben classificata in questa nuova classificazione di WHO. E poi ci sono anche delle leucemie acute mieloidi non meglio caratterizzate, cioè ancora ci sono delle entità nosografiche che non si riesce bene ad incasellare in queste classificazioni, ecco perché poi le classificazioni cambiano progressivamente man mano che abbiamo informazioni migliori soprattutto su quelle che sono le alterazioni molecolari che sottendono a questa modificazione neoplastica. Qual è l'approccio terapeutico che noi utilizziamo nei pazienti con leucemia acuta? L'approccio terapeutico è quello di fare una chemioterapia aggressiva con due farmaci o tre farmaci, usualmente utilizziamo una antraciclina più la citosina-arabinoside e questa terapia molto aggressiva, perché determina un temporaneo azzeramento del midollo osseo e quindi un azzeramento dei valori di produzione dei globuli bianchi, delle piastrine, dei globuli rossi e quindi un rischio di infezione, un rischio di emorragie, comunque grossomodo questo determina una remissione completa intorno al 75%, dove per remissione completa intendiamo la scomparsa di quelle famose cellule blastiche che abbiamo visto all'esordio di malattia. Successivamente però bisogna consolidare questa risposta e si fa appunto una intensificazione sempre utilizzando chemioterapia in particolare si utilizzano alti dosaggi di ara-ci ma ci sono altri schemi che si utilizzano in altre condizioni. A quel punto bisogna scegliere se continuare con la chemioterapia, se fare un autotrapianto o fare un trapianto allogenico e questa scelta dipende da quello che noi abbiamo definito nella fase diagnostica di questa malattia, cioè soprattutto dipende dalle alterazioni citogenetiche che presenta il paziente: - se questo paziente presenta delle alterazioni citogenetiche favorevoli allora può fare anche la semplice chemioterapia, - se presenta delle alterazioni citogenetiche sfavorevoli deve fare il trapianto allogenico, ovviamente se è nelle condizioni di farlo, se è un paziente relativamente giovane, se non ha grosse comorbilità, bisogna cercare un donatore compatibile, ci sono tutte delle limitazioni e variabili, - oppure l'alternativa è quella di fare un auto trapianto, cioè sfruttare le proprie cellule staminali per poter fare un dosaggio elevato di chemioterapia, in questo modo nei pazienti di età inferiore ai 60 anni si ottiene una percentuale di guarigione o di long term overall survival che è intorno al 35%. Quindi per il momento questi sono i numeri, noi riusciamo a guarire circa il 35% dei pazienti giovani, inferiore ai 60 anni. E se andiamo a guardare le curve basate su tutti i pazienti senza fare distinzione tra basso rischio, alto rischio ecc, grossomodo vediamo che non c’è differenza nelle curve di sopravvivenza fra i pazienti che fanno l’autologo, i pazienti che fanno l’allogenico e i pazienti che fanno soltanto alte dosi di chemioterapia. Questa curva di sopravvivenza deriva dal fatto che il trapianto allogenico è sicuramente il più efficace in termini di controllo della malattia, ma è anche quello che determina la maggiore mortalità per cui questa maggiore mortalità va a compensare la maggiore efficacia. Il trapianto autologo e le alte dosi di chemioterapia sono sicuramente meno attive nei confronti della malattia in termini di efficacia ma siccome non determinano mortalità allora questo compensa la loro minore efficacia. Alla fine in termini di sopravvivenza globale, le tre opzioni terapeutiche si equivalgono. Tutto questo invece non vale per i pazienti anziani dove le curve di sopravvivenza sono veramente sconfortanti e negli anni non c’è stato nessun miglioramento: si è cercato in tanti modi di modificare e di migliorare la terapia; ci sono tre schemi terapeutici differenti ma i risultati sono assolutamente sovrapponibili, nei pazienti anziani, di età >60 anni la probabilità di ottenere una lunga sopravvivenza è meno del 20%. Un’ altra possibilità terapeutica, oltre alla chemioterapie e al trapianto, anche per le leucemie mieloidi, malattie mieloidi, esistono gli anticorpi monoclonali, e in particolare esiste un anticorpo monoclonale anti CD33 che ha una peculiarità: intanto il CD33 è espresso nel 90% delle cellule di leucemia acuta mieloide ma non è espresso sulle cellule staminali emopoietiche, quindi è un trattamento ideale sul piano teorico per questi pazienti. La cosa interessante è che questo anticorpo anti CD33 viene coniugato con un farmaco che si chiama calicheamicina, che è in pratica una antraciclina, e quindi l’anti CD33 non fa altro che veicolare nel bersaglio cellulare questa sostanza che è fortemente citotossica. Il meccanismo d’azione di questo farmaco è quello di legarsi al CD33, internalizzarsi, liberare dentro la cellula la calicheamicina, la quale è in grado di intercalarsi con la molecola a doppia elica del DNA e indurre apoptosi. In realtà questo schema teorico e assolutamente interessante finora non ha dato risultati del tutto brillanti, quindi è un farmaco che viene utilizzato in associazione alla chemioterapia ma non ha un peso così importante com’è stato invece il peso che hanno dimostrato gli anticorpi monoclonali nelle malattie linfoproliferative. LEUCEMIA ACUTA PROMIELOCITICA Un sottotipo di leucemia acuta mieloide è la cosiddetta leucemia acuta promielocitica o M3 della classificazione FAB. E’ interessante approfondirne la conoscenza perché questa leucemia è passata dall’essere la leucemia acuta peggiore, quella a prognosi più sfavorevole, è diventata quella a prognosi più favorevole e poi ha delle peculiarità: - quasi sempre si associa ad una cosiddetta coagulazione intravascolare disseminata cioè ad un disordine coagulativo nel quale c’è un eccesso di coagulazione, si chiama coagulazione intravascolare disseminata, eccesso di coagulazione che provoca un consumo dei fattori della coagulazione e di conseguenza una sindrome emorragica. Quindi questa è una condizione particolarmente difficile perché è una condizione emorragica che dipende da un eccesso di coagulazione, quindi si capisce bene che non è una cosa facile da trattare o facile da gestire. - Inoltre questa malattia, contrariamente alla maggior parte delle leucemie acute si manifesta con leucopenia e non con leucocitosi, generalmente nelle leucemie acute noi abbiamo un aumento dei blasti circolanti, quindi uno dei segnali che noi vediamo nel sangue periferico è l’aumento dei globuli bianchi, nel caso della leucemia acuta promielocitica vediamo una leucopenia, più spesso, non sempre. - Poi c’è una tipica associazione con la t(15;17) che è patognomonica della leucemia acuta promielocitica: questa traslocazione provoca la formazione di un trascritto ibrido che si chiama PMLRARα, che permette a questa leucemia di essere sensibile a dosi elevate di acido oltransretinoico che è un derivato della vitamina A, oltre ad essere sensibile anche all’arsenico triossido. Questo è l’aspetto morfologico tipico delle leucemia promielocitica dove si vedono questi bastoncelli nel citoplasma che si chiamano corpi di Auer, trovare queste macchie di corpi di Auer è un elemento morfologico assolutamente distintivo e patognomonico delle leucemia acuta promielocitica, non c’è bisogno di fare nessuna indagine citogenetica e molecolare, una volta che si vedono questi aspetti morfologici questa è una tipica leucemia acuta promielocitica. Purtroppo non sempre è così perché ci sono delle varianti di leucemia acuta promielocitica che sono di difficile interpretazione morfologica, però se uno vede un quadro di questo genere la diagnosi è fatta. Si deve fare però la valutazione citogenetica e FISH che ci permette appunto di vedere questa tipica traslocazione cosiddetta PML-RARα, cioè la formazione di un trascritto molecolare ibrido che mette insieme il gene pml col gene rar alfa. Quindi su un piano clinico dicevamo che questa malattia si accompagna ad una grave sindrome emorragica che dipende da una coagulazione intravascolare disseminata e quindi questi pazienti hanno delle gravi soffusioni emorragiche ma soprattutto hanno delle gravi emorragie cerebrali e questo è molto importante perché questa malattia è abbastanza improvvisa nella sua espressione: quei casi di leucemia acuta fulminante nella maggior parte dei casi sono leucemie acute promielocitiche; è tipica la descrizione che fece Jean Bernard alcuni anni fa di un ciclista che vinse la sua tappa al giro di Francia e il giorno dopo gli è stata diagnosticata la leucemia acuta promielocitica, e non è raro fare diagnosi di LAP in pazienti in rianimazione per emorragie cerebrali: quindi è una malattia che costituisce una delle poche emergenze ematologiche dove bisogna intervenire subito perché il rischio emorragico è tale che questi pazienti possono morire per l’emorragia in maniera immediata. Quindi questa è tipicamente la traslocazione t(15;17) che è patognomonica della lap e che determina la formazione di un trascritto ibrido che si chiama pml-rar alfa. Questo è importante perché permette di capire il meccanismo di azione delle vitamina A: dicevamo che questa leucemia è l’unica sensibile all’acido oltransretinoico (derivato della vitamina A). Questa è un’informazione che ci è venuta dalla medicina cinese: i cinesi hanno scoperto che trattando questi pazienti con l’acido oltransretinoico, questi andavano in remissione, non avevano capito il motivo ma adesso si è capito, e dipende dal fatto che in condizioni normali il recettore per l’acido oltransretinoico è legato a un meccanismo di repressione e questo meccanismo impedisce la trascrizione del DNA e la maturazione della cellula; l’azione dell’acido retinoico permette di liberare questo sistema e di liberare la DNA-polimerasi, per cui una quantità fisiologica di acido retinoico, quella che noi abbiamo nel nostro organismo (che è la vitamina A), permette di liberare questa DNA polimerasi e quindi permette il normale differenziamento cellulare, se invece non c’è l’acido retinoico questo differenziamento cellulare non avviene, quindi dosi fisiologiche di acido retinoico permettono la normale maturazione delle cellula. Quando invece c’è la traslocazione t(15;17) che lega i due geni, rar alfa con pml, succede che questo complesso trascrizionale è molto più legato, è molto più coeso, quindi il pml non fa altro che fungere da collante tra questo complesso trascrizionale e il gene per il recettore dell’acido retinoico. Quindi in condizioni normali, la quantità normale di acido retinoico che abbiamo non riesce a forzare questo meccanismo e quindi ci vogliono dosi più elevate di acido retinoico che è quello che diamo farmacologicamente e quindi se diamo una dose più elevata di a.r. questo forza il sistema e permette alla DNA polimerasi di agire e il differenziamento cellulare. Se noi prendiamo queste cellule di lap e mettiamo dentro l’acido retinoico permettiamo un differenziamento delle cellule. (slide) Vediamo la curva di sopravvivenza dei pazienti con lap che venivano trattati prima di questa scoperta e vediamo la curva di sopravvivenza dei pazienti con lap trattati con aor. In realtà il gruppo italiano e spagnolo hanno combinato insieme la chemioterapia e l’aor e ha ottenuto dei risultati strabilianti: vediamo una curva di sopravvivenza di pazienti affetti da lap a basso rischio che è una curva di sopravvivenza del 100%, c’è da dire che è una curva un po’ ottimistica perché mancano tutti quelli che sono morti subito, quei pazienti che per cause emorragiche hanno avuto una morte immediata però fondamentalmente una volta superata questa fase, che è la fase più rischiosa per questi pazienti, i pazienti rispondono molto bene al trattamento con aor e hanno una curva di sopravvivenza che è la migliore delle curve per le leucemie acute. Domanda di un collega: ‘Quindi prof, l’acido retinoico sbloccando il meccanismo, che succede? Come guariscono i pazienti? Il clone va in differenziamento o va in apoptosi?’ Prof: ‘Tutti e due! Il clone prima si differenzia e poi va in apoptosi; se poi ci aggiungi la chemioterapia aumentiamo l’apoptosi ed eliminiamo le cellule immature che non sono riuscite ad andare in differenziamento e apoptosi per opera dell’acido retinoico da solo.’ Quei pochi pazienti che non rispondono all’acido retinoico rispondono all’arsenico che ha un meccanismo di azione abbastanza simile. LEUCEMIA ACUTA LINFOBLASTICA Caratteristiche La leucemia acuta linfoblastica (lal) è una leucemia che occorre in circa il 20 % dei pazienti adulti con leucemia acuta. E’ una leucemia che tipicamente interessa però i bambini, picco di incidenza per età: intorno ai 4 anni, fino a 4anni 7 per 100000 bambini da 5 a 9 anni 3-4 per 100000 poi si riduce drasticamente da 15 anni in su è soltanto 1 per 100000 poi diventa 0.4-0.6 e per età > 60 anni c’è un lievissimo aumento di nuovo della frequenza. Quindi questa è la curva di incidenza della leucemia acuta linfoblastica, col picco nell’età infantile e poi una netta regressione di incidenza e un lieve aumento nell’età anziana. (slide) Se questa è la curva di incidenza della leucemia acuta linfoide, questa è la curva di mortalità della leucemia acuta linfoide e già queste due diapositive vi fanno capire qual è la modalità di trattamento di queste due forme: elevata incidenza in età infantile, elevata mortalità nell’età adulta, questo vuol dire che nei bambini noi riusciamo a far guarire quasi tutti questi pazienti affetti da leucemia acuta linfoide, negli adulti non riusciamo a far guarire nessuno, negli anziani soprattutto, quindi vediamo questa netta differenza fra l’incidenza e la mortalità. Sintomatologia I segni della LAL sono non specifici: - febbre - sanguinamento dolori ossei linfadenopatia dolori muscolo-scheletrici aumento delle linfoadenopatie, essendo una malattia linfoproliferativa ci possono essere segni di localizzazione meningea di malattia: la leucemia acuta linfoblastica ha una peculiare tendenza a localizzarsi al SNC, in particolare nelle meningi piuttosto che nel parenchima cerebrale, noi parliamo in questo caso di meningosi leucemica che si può manifestare appunto con i segni dell’ipertensione endocranica, cefalea vomito ecc. Un’altra sede peculiare di localizzazione è la localizzazione testicolare che è abbastanza tipica della LA Linfoblastica mentre è una rarità nella LA Mieloide; e poi come tutte le malattie linfoproliferative può essere interessato il mediastino, ci possono essere masse mediastiniche e molto raramente possono dare una sindrome tipo sindrome da compressione della vena cava superiore. Chiaramente l'aspetto diagnostico più importante si ha a carico dell'emocromo: i pazienti sono spesso anemici, piastrinopenici, anche se la piastrinopenia non sempre è una piastrinopenia grave, circa il 50% dei pazienti ha dei sintomi di sanguinamento e poi spesso c'è, anzi quasi sempre c'è un aumento dei globuli bianchi che però non è un aumento significativo, più spesso sono pazienti che hanno un numero di globuli bianchi < 10000 talvolta > 50000. Immagine: bambino con manifestazioni emorragiche che sono indicative di leucemia acuta e poi si vede un mediastino ingrossato in un paziente con una leucemia acuta a cellule T; si vede poi una localizzazione testicolare. Classificazione Su un piano morfologico le Leucemie Acute Linfoidi hanno anch’essi una classificazione FAB sempre Franch-American-British e però si distinguono solo tre tipi di leucemia acuta linfoblastica sul piano morfologico: L1, L2 e la L3. La L1 è caratterizzata da questi linfociti piccoli, con scarso citoplasma con cromatina addensata, si vedono tutti uguali sul piano morfologico; la L2 invece è molto più pleomorfa e caratterizzata da questi granuli con maggiore citoplasma, con cromatina più lassa, la presenza di questi nucleoli; la L3 è caratterizzata da tipici vacuoli citoplasmatici che ne fanno una caratteristica morfologica peculiare configurando questo L3 la forma leucemica del linfoma di Burkitt cioè queste stesse cellule se li troviamo nel linfonodo, se non li troviamo nel sangue periferico, questa malattia la chiamiamo linfoma di Burkitt, se invece le troviamo nel sangue periferico questa malattia si chiama leucemia acuta linfoide L3. Essendo dei linfociti chiaramente queste cellule possono essere soggette ad una valutazione in citofluorometria che ci permette di differenziare il grado di maturazione, per cui nella maggior parte dei casi si tratta di linfociti che appartengono alla linea B, più raramente linfociti che appartengono alla linea T, e nella maggior parte dei casi i linfociti che appartengono alla linea B sono linfociti cosiddetti precursori B o pre-B, mentre più rari sono i linfociti maturi che appartengono alla linea B matura e che sono quegli stessi che abbiamo visti prima e che configurano la L3 o il linfoma di Burkitt. Anche qui ci sono delle alterazioni citogenetiche importanti, in particolare ci possono essere delle leucemie acute con traslocazione t(9;22) che è una traslocazione tipica della leucemia mieloide cronica ma che può essere presente anche nella leucemia acuta linfoide, questo cromosoma (9;22) è il cosiddetto cromosoma Philadelphia, che è la città dove per prima venne riconosciuto. Ma ci sono tutta una serie di altre possibili alterazioni, ricorre di nuovo quel gene MLL che abbiamo visto per le leucemia acute mieloidi, ci sono altre alterazioni che interessano altri geni e altri cromosomi. Prognosi Quindi anche qui nel caso della leucemia acuta linfoide dobbiamo definire le caratteristiche prognostiche di questi pazienti perché questo è importante per l’aggressività della terapie quindi le caratteristiche prognostiche di questi pazienti sono: l’età e la presentazione clinica: abbiamo visto che è importante l’età, i bambini guariscono e gli anziani no; le caratteristiche biologiche della malattia, l’importanza dell’alterazione citogenetica un altro parametro con un valore prognostico molto importante ma che si valuta solo dopo la terapia è la risposta iniziale alla terapia. Quindi i pazienti cosiddetti rischio standard, che hanno un rischio non elevato, sono i pazienti che hanno: - una bassa età <35anni, - i pazienti con bassa conta di globuli bianchi <30000 per quanto riguarda la linea B e <100000 per quanto riguarda la linea T, - i pazienti la cui leucemia appartiene alla linea T e non alla linea B, - i pazienti che ottengono la remissione totale, cioè la scomparsa della malattia entro 4 settimane. Ma è molto importante il valore della citogenetica e la citogenetica è probabilmente uno dei motivi più importanti perché i bambini vanno meglio rispetto agli adulti, infatti se prendiamo uno dei parametri prognostici peggiori che è quello del cromosoma Philadelphia t(9;22), vedete come nei bambini la percentuale di pazienti che hanno questa traslocazione è molto più bassa rispetto agli adulti, quindi sono due malattie, quella dei bambini e quella degli adulti che sono biologicamente differenti e quindi queste traslocazioni cromosomiche (4;11), (9;22), (13;21), sono differentemente presenti nei bambini e negli adulti: per esempio la (9;22) solo nel 3% dei bambini è presente, 20% degli adulti, 40-50% degli anziani; viceversa la (4;11) che è una alterazione citogenetica più favorevole è presente nel 50% dei bambini ed è quasi assente negli adulti e viceversa. Quindi abbiamo pazienti che presentano caratteristiche citogenetiche favorevoli e pazienti che presentano caratteristiche citogenetiche sfavorevoli, esattamente come succede per la leucemia acuta mieloide: quindi anche per la leucemia acuta linfoblastica è importante valutare bene il cariotipo prima di iniziare il trattamento perché questo ci deve guidare poi nell’intensità del trattamento. Trattamento Il trattamento che facciamo in questi pazienti è un trattamento che consiste in una fase di induzione che è abbastanza simile a quella che utilizziamo per la leucemia acuta mieloide, una fase di consolidamento e poi in maniera molto differente rispetto alla LAM un mantenimento che dura addirittura tre anni, cioè sono pazienti che per tre anni fanno una terapia di mantenimento con basse dosi di chemioterapici. Accanto a questo bisogna fare la cosiddetta profilassi del SNC, perché come abbiamo detto prima in questa malattia c’è una spiccata tendenza di queste cellule di andare a indovarsi del SNC, in particolare nelle meningi: le meningi appartengono al SNC e come sappiamo esiste la cosiddetta barriera ematoencefalica: questa impedisce ai farmaci che noi utilizziamo per il trattamento della leucemia di passare dal sangue nel cervello, e quindi queste cellule crescono indisturbate, si dice che il SNC costituisce un “santuario” per le cellule di LAL, nel senso che lì le cellule crescono e nessuno le disturba, perlomeno i chemioterapici fatti per via endovenosa o per bocca non sono i grado di arrivare a quel livello. Ecco perché occorre fare le cosiddette punture lombari medicate, cioè iniettare nello speco intratecale dei farmaci chemioterapici e lo si fa a scopo di profilassi per impedire che ci possa essere una localizzazione di questa malattia a livello del SNC. Tutta questa chemioterapia che facciamo e anche la profilassi del SNC (si usava la cosiddetta irradiazione craniale, cioè l’irradiazione dell’encefalo proprio per impedire la localizzazione di questa cellule a livello del SNC), queste condizioni portano degli effetti collaterali: intanto ci sono degli effetti collaterali legati ai chemioterapici, c’è la cosiddetta sindrome da lisi tumorale che è tipica della LAL, cioè queste cellule hanno una così elevata sensibilità al trattamento che la lisi di queste cellule è così rapida che determina la liberazione di alcune sostanze tossiche per l’organismo, in particolare ci può essere una ipercaliemia che può essere mortale perché determina un arresto cardiaco; poi ci possono essere tutte le conseguenze della chemioterapia quali il sanguinamento, le infezioni, le mucositi ecc. Effetti collaterali del trattamento Ma è importate sottolineare il fatto che ci possono essere degli eventi avversi tardivi, questi eventi tardivi sono soprattutto legati ad un eccesso di uso di radioterapia craniale che determina una compromissione del SNC, nei bambini ci può essere un ritardo della crescita, la chemioterapia può indurre cardiotossicità, può indurre infertilità e tutti i chemioterapici possono indurre incidenza di seconde neoplasie. può succedere quindi che pazienti con una LAL, bambini con LAL che guariscono per la LAL poi sviluppano un’altra neoplasia! (slide) E questo è appunto in una casistica a lungo termine di bambini guariti per LAL, vedete come il rischio di sviluppare una nuova neoplasia a 14-15 anni, soprattutto nei pazienti che hanno subito la radioterapia craniale come profilassi o terapia della meningosi leucemica. Ma accanto a questi ci può essere neurotossicità, che è legata sempre alla irradiazione craniale, alle alte dosi di chemioterapici che passano la BEE (barriera emato-encefalica), alle alte dosi di desametasone che si utilizzano in questi casi e alla terapia intratecale che si utilizza appunto per la profilassi o per la terapia della meningosi leucemica. Questi pazienti fanno una terapia di mantenimento che due 2-3 anni e tutto questo serve per eliminare la malattia minima residua. Valutazione della malattia minima residua Valutare dopo la terapia quanto residuo di malattia c’è rappresenta la nuova frontiera del trattamento delle leucemie acute linfoblastiche: infatti nei bambini troviamo che al momento della remissione, cioè al momento in cui non vediamo più cellule leucemiche, andando a valutare la malattia minima residua con metodiche molecolari, quasi tutti i bambini hanno una percentuale di malattia minima residua che è 0.01%: questi bambini hanno un’eccellente prognosi. se invece questi bambini hanno una quantità di residuo molecolare che dello 0.1%, quindi dieci volte di più, in questo caso hanno un elevato rischio di ricaduta. Questo discorso vale per i bambini ma vale ancor di più per gli adulti: se noi andiamo a fare la valutazione della malattia minima residua ad un anno dal trattamento, le persone che hanno una piccola malattia residua hanno una percentuale di sopravivenza che è maggiore di quella dei pazienti che hanno una malattia minima residua più importante. Quindi sicuramente abbiamo avuto negli ultimi 20 anni un miglioramento di quelli che sono stati i fattori di rischio che sono l’età e il numero dei globuli bianchi, le caratteristiche biologiche (per esempio la citogenetica e il fenotipo), e la risposta al trattamento: pazienti che rispondono più precocemente sono quelli che hanno una probabilità maggiore di guarire. Chiaramente tutto questo deve essere valutato come risposta molecolare. Protocolli pediatrici vs protocolli per gli adulti Un punto importante da sottolineare è che in effetti forse non è soltanto una differenza biologica quella che fa differire la prognosi tra bambini e adulti: noi sappiamo che c’è un’alta remissione sia nei bambini che negli adulti però poi se andiamo a vedere la percentuale di leukemia-free survival è molto più elevata nei bambini che negli adulti: al primo impatto, alla prima terapia non è che vanno in remissione soltanto i bambini, o meglio i bambini ci vanno con maggiore percentuale (il 97% dei bambini va in remissione contro l’80-90% degli adulti), però poi è la sopravvivenza libera da malattia quella che poi fa la differenza, cioè gli adulti vanno pure in remissione, ma poi ricadono mentre i bambini vanno in remissione e la mantengono. (slide) questa è la curva di sopravvivenza della leucemia acuta dei bambini fino al 1966, questa è quella fino al 1979, questa è fino al 1983, questa è fino al 1991 e questa è fino al 1997: vedete come in 30 anni di trattamento della LAL c’è stato un significativo miglioramento della capacità di guarire questi pazienti. Il trattamento della LAL del bambino è il paradigma dei successi della terapia antineoplastica in generale, cioè se noi avessimo questi successi in tutte le neoplasie saremmo ben felici, purtroppo ce l’abbiamo soltanto in poche neoplasie; vedete come si è passati da una capacità di sopravvivenza che è meno del 10%, ad una probabilità di sopravvivenza che è del 90%. Questo grazie a tutti i successi che si sono ottenuti, perché man mano si è capito meglio come utilizzare questi farmaci e soprattutto si è capito meglio la biologia e la fisiopatologia della malattia e quindi quando piazzare questi farmaci. Purtroppo questo non succede negli adulti, dove ci sono stati dei miglioramenti negli anni, ma vedete come nei pazienti di età superiore ai 40 anni la probabilità di ottenere una buona guarigione si aggira intorno al 20%, quindi siamo ben lontani da quel 90% dei bambini. E’ vero che anche negli adulti c’è stato un miglioramento, infatti se consideriamo il quadriennio dall’80 all’84, e il quadriennio dal 2000 al 2004, vedete come la percentuale di miglioramento in termini di sopravvivenza c’è stata in tutte le fasce di età tranne nei pazienti di età superiore ai 60 anni, ma già in questi pazienti dai 15 ai 60 anni c’è stato un significativo aumento della probabilità di sopravvivenza dal 14 al 20%. E’ vero come dicevamo prima che i bambini hanno una malattia differente rispetto agli adulti, nel senso la percentuale di bambini che hanno una citogenetica caratterizzata dal cromosoma Philadelphia varia dall’1 al 3%. Viceversa se vediamo tutti protocolli degli adulti la percentuale di pazienti affetti da LAL con cromosoma Ph positivo, vanno dal 17 al 24%, quindi sicuramente i bambini hanno meno alterazioni cromosomiche sfavorevoli rispetto a quelle degli adulti; inoltre, se prendiamo il numero di globuli bianchi, che è un’altra caratteristica prognostica sfavorevole, vedete come la mediana di globuli bianchi all’esordio di questi bambini è intorno ai 10-12000gb, viceversa la mediana di globuli bianchi all’esordio degli adulti è intorno a 20-30000gb. Quindi non c’è dubbio che c’è una differenza in termini biologici tra la LAL del bambino e quella dell’adulto, però c’è un aspetto: se si prendono gli adolescenti, cioè quelli che stanno tra 15 e 20 anni, si tratta di una fascia di pazienti che a seconda se vanno a finire in un reparto di ematologia dell’adulto o in reparto di ematologia pediatrica, vengono trattati in maniera differente: se vano a finire in pediatria vengono trattati con dei protocolli pediatrici, se vanno a finire in ematologia dell’adulto vengono trattati con protocolli dell’adulto. Quando si è andati a confrontare l’outcome di questi pazienti trattati con protocolli pediatrici o con protocolli degli adulti, in uno studio francese si è visto per esempio che questi pazienti avevano il 78% di sopravvivenza a 5 anni se trattati con protocollo pediatrico, e invece del 45% se trattati con protocollo degli adulti. Lo stesso per questo studio americano: 67% a 7 anni se trattati cono protocollo pediatrico, 46% a 7 anni se trattati con un protocollo dell’adulto. E questi sono altri studi che non valutano la sopravvivenza globale ma valutano la cosiddetta event free survival, cioè la sopravvivenza libera da eventi, ma che dicono tutti quanti la stesa cosa: gli adolescenti trattati con un protocollo pediatrico vanno molto meglio rispetto agli adolescenti trattati con protocollo dell’adulto: quindi la nuova tendenza è di trattare questi pazienti adolescenti o di trattare anche gli adulti con protocolli diciamo copiando i protocolli pediatrici. Cosa hanno di differente i protocolli pediatrici rispetto a quelli dell’adulto? L’elemento che hanno di maggiore differenza è l’uso di un farmaco che si chiama L-asparaginasi . L’asparaginasi è un farmaco che impedisce la formazione di asparagina, cioè impedisce alla asparagina di entrare dentro la cellula, e siccome la cellula leucemica non ha la capacità di formare asparagina (non ha asparagina sintetasi), la cellula leucemica muore affamata, perché le impediamo di avere la asparagina, che è un aminoacido essenziale. Una delle differenze fondamentali che esiste tra i protocolli pediatrici e quelli dell’adulto è appunto l’utilizzo nei protocolli pediatrici di dosaggi di L-asparagina 10 volte più alti di quelli dell’adulto, e perché questo? Perché l’asparaginasi è un farmaco tossico, nell’adulto in particolare è un farmaco che dà delle disfunzioni epatiche e che dà delle disfunzioni dell’emostasi, può determinare delle gravi emorragie ma anche e soprattutto delle gravi trombosi e può dare delle gravi pancreatiti per cui nell’adulto è molto più difficile utilizzare questo farmaco rispetto al bambino. (slide) Questo è uno studio interessante che utilizza l’asparaginasi e che ha fatto una valutazione: dal momento che la asparaginasi può provocare trombosi, i pazienti nei quali non c’è stata la trombosi vanno molto meglio rispetto ai pazienti in cui c’è stata la trombosi. Allora l’interpretazione di questo studio qual è? Non è che la trombosi che ha portato a morte, ma la trombosi è stata quella che ha portato ad una riduzione del dosaggio di asparaginasi (proprio perché è quella che provoca la trombosi) e allora utilizzare meno asparaginasi significa dare una minore chance di guarigione a questi pazienti. Una spiegazione alternativa potrebbe essere che siccome appunto questi pazienti hanno avuto trombosi, sono stati costretti ad interrompere il trattamento e quindi non si è rispettata quella cosiddetta intensità di dose che è necessaria per il trattamento di malattie che sono a così rapida attività proliferativa: comunque la vogliamo mettere il problema rimane sempre lo stesso, cioè l’impossibilità a causa degli effetti collaterali di poter condurre una terapia a dosaggio più elevato possibile e nei tempi più rapidi possibili. E infine il problema del trapianto nella LAL soprattutto degli adulti, perché la terapia negli adulti risulta insoddisfacente e anche se conosciamo meglio la malattia e i fattori prognostici, il trapianto allogenico sicuramente è più efficace almeno nei pazienti a cattiva prognosi. E’ stato fatto uno studio che ha confrontato, in pazienti che avevano ottenuto una risposta alla chemioterapia, l’efficacia del trapianto allogenico oppure dell’autotrapianto, oppure della chemioterapia (erano tutti pazienti di età <55anni, non affetti da leucemia acuta Ph+, perché per i pazienti Philadelphia positivi c’è una terapia completamente differente), questi pazienti erano in remissione completa dopo una chemioterapia di induzione: - a quel punto i pazienti che avevano il donatore compatibile, cioè che avevano il parente, fratello o sorella, compatibile per l’HLA, venivano sottoposti ad un’altra chemioterapia e poi dopo al trapianto allogenico; - gli altri, quelli che non ce l’avevano, venivano sottoposti ad un’altra chemioterapia del tutto simile e poi randomizzati a ricevere un autotrapianto oppure una terapia standard con un mantenimento per altri due anni e mezzo. * Donatore significa che ha fatto il trapianto allogenico, o per lo meno l’intenzione era di andare a fare il trapianto allogenico, poi per qualche motivo magari non l’ha fatto, questa analisi si fa in cosiddetta intention to treat. La sopravvivenza dei pazienti ad alto rischio, nei pazienti che avevano il donatore era sicuramente più elevata dei pazienti che non avevano il donatore, sia nell’alto rischio che nel basso rischio: questa differenza non è statisticamente significativa, mentre è statisticamente significativa la differenza di ricaduta, perché se vogliamo andare a guardare l’efficacia soltanto dobbiamo guardare la ricaduta, se vogliamo guardare la sopravvivenza in generale dobbiamo guardare anche la tossicità. La differenza in termini di ricaduta è netta ed è statisticamente significativa, a favore di chi aveva il donatore, quindi a favore di chi ha fatto il trapianto allogenico, questo vuol dire che in termini di efficacia antileucemica il trapianto allogenico è sicuramente più efficace rispetto all’autotrapianto o alla chemioterapia convenzionale, anche se poi questo effetto così netto in termini di ricaduta è un po’ meno netto in termini di sopravvivenza globale perché qui pesa anche la tossicità, la mortalità legata al trapianto allogenico. La sorpresa è stata che la chemioterapia va meglio del trapianto autologo, per cui questo è uno studio che ha dato un colpo di grazia a quello che era già la vacillante valutazione del trapianto autologo nella leucemia acuta linfoblastica e adesso la maggior parte dei protocolli chemioterapici prevedono che nei pazienti ad alto rischio si faccia o si tenti un trapianto allogenico, mentre nei pazienti a basso rischio si può fare anche una chemioterapia convenzionale. Lezione 13 Le MIELODISPLASIE Nelle mielodisplasie noi abbiamo intanto anomalie quantitative, si verifica infatti una citopenia periferica che può essere pan-, bi- o monolineare. Si può verificare dunque anemia, leucopenia o piastrinopenia, queste tre condizioni possono comparire tutte contemporaneamente o se ne può verificare una sola oppure due in combinazione. L’anemia è caratterizzata un volume globulare medio normale nel 40% dei casi, aumentato nel 50% dei casi e nel 10% dei casi può essere diminuito. L’RDW o indice di anisocitosi aumentato, per anomalie della curva di distribuzione volumetrica. Il numero di reticolociti è normale o diminuito e raramente si verifica un loro aumento. Ci può pure essere leucopenia che è soprattutto neutropenia ma ci possono essere anche condizioni di leucositosi con monocitosi. Infine ci può essere piastrinopenia con MPV normale o aumentato. La mielodisplasia è caratterizzata da un’emopoiesi inefficace. Nel midollo possiamo trovare una cellularità normale oppure aumentata, qualche volta può essere anche diminuita. In alcuni casi si verifica un’emofagocitosi cioè di macrofagi che inglobano i globuli rossi. Spesso nel midollo si ha un aumento dei depositi marziali e ci possono essere i cosiddetti sideroblasti ad anello (eritroblasti in cui il ferro si dispone come un anello intorno al nucleo). Ci possono essere anche blasti, cioè cellule neoplastiche (si riconoscono al microscopio per i corpi di Auer che rappresentano una indicazione abbastanza patognonomica), sia nel midollo che nel sangue periferico. Soprattutto si ha una displasia morfologica con alterazioni morfologiche degli elementi, da cui il nome mielodisplasia. Definizione E’ una malattia dell’anziano e l’incidenza aumenta con l’età, dai sessanta agli ottantacinque anni ed oltre. Si tratta di malattie clonali e quindi neoplastiche, delle cellule staminali ematopoietiche. Sono situazioni patologiche caratterizzate da un’emopoiesi inefficace, cioè il midollo viene infatti trovato a cellularità normale o aumentata però troviamo poi una citopenia periferica. Questa discrepanza fra l’attività mielopoietica, midollare e la citopenia periferica si chiama ematopoiesi inefficace, perché il midollo lavora ma non riesce a produrre normali quantità di elementi. Troviamo inoltre vari dismorfismi (displasie morfologiche) nelle filiere maturative. Nel sangue periferico si verifica una citopenia che può essere pan-, bi- o monolineare e si ha inoltre una variabile tendenza all’evoluzione in leucemia acuta. Ricordate che si tratta sempre di malattie clonali e dunque neoplastiche anche quando non evolvono in leucemia. Dal momento che si tratta di patologie molto eterogenee (come visto ci può essere neutropenia ma a volte anche una monocitosi, citopenia mono, bi-, trilineare etc), allora si è cercato di classificare queste mielodisplasie che sono piuttosto sfuggenti alla classificazione. Però è stata fatta una classificazione nel 1976 e poi confermata nel 1982 che è la classificazione FAB, basata esclusivamente su elementi morfologici e biochimici. Era una classificazione semplice e riproducibile che distingueva forme a basso ed alto rischio di trasformazione leucemica. E’ stata una classificazione che ha tenuto per molto tempo ma poi è stata modificata e in parte sostituita sulla spinta delle critiche. Comunque questa classificazione vale ancora oggi e può essere utilizzata. Questa classificazione permette di inquadrare diverse malattie, che sono: - anemia refrattaria - anemia refrattaria con sideroblasti ad anello (il ferro si dispone fondamentalmente nei mitocondri a formare una sorta di anello attorno al nucleo L’anemia refrattaria e l’anemia refrattaria con sideroblasti ad anello sono condizioni a basso rischio. Poi ci sono le forme ad alto rischio che sono caratterizzati dalla presenza di blasti leucemici, ovvero si sono già identificate morfologicamente delle cellule fortemente atipiche che configurano quelle condizioni a rischio di evoluzione verso una leucemia acuta mieloide. - anemia refrattaria con eccesso di blasti (percentuale di blasti a livello midollare tra 6 e 19%) - anemia refrattaria con eccesso di blasti in trasformazione leucemica (percentuale di blasti compresa tra il 20% ed il 30%) Questa era quindi la vecchia classificazione FAB, molto semplice ma ancora valida perché permetteva di fare una classificazione che avesse un risvolto prognostico. Si chiamano anemie refrattarie perché in passato erano refrattarie a qualsiasi terapia convenzionale (ferro, vitamina B12, acido folico) in quanto non sono anemie da carenza ma si tratta di un’alterazione neoplastica che rende l’eritrone poco funzionante. CLASSIFICAZIONE FAB Questa classificazione venne però criticata perché non permetteva di classificare alcuni quadri patologici, tra cui la mielodiplasia con fibrosi midollare, la mielodisplasia ipocellulare, la mielodisplasia con assenza di anemia ma presenza di piastrinopenia e/o leucopenia e le mielodisplasie in età pediatrica ma soprattutto perché per motivi storici nella classificazione FAB manca il fondamentale ed indiscutibile apporto della citogenetica. Anche qui ancora una volta la citogenetica sta diventando una delle valutazioni biologiche necessarie per fare un corretto inquadramento della mielodisplasia perché anche qui la citogenetica ha una valore prognostico. Inoltre anche sulla percentuale di blasti non c’era accordo perché in molti ritenevano che una percentuale di blasti (blastosi) superiore al 20% corrispondesse già ad un quadro di leucemia acuta. Da queste osservazioni, mentre nella classificazione FAB tra il 20 e il 30% di blasti si parlava di anemia refrattaria con eccesso di blasti in trasformazione leucemica, adesso già con più del 20% di blasti si parla direttamente di leucemia acuta, ovvero il cut-off di distinzione per le mielodisplasie viene considerato soltanto il 10%. Questo significa che nella nuova classificazione è stata abolita la RAEB-T (anemia refrattaria con eccesso di blasti in trasformazione leucemica), la leucemia mielomonocitica cronica è stata trasferita in altra categoria ed è stata considerata a parte un’alterazione citogenetica specifica che è la sindrome del 5q – (leggi “sindrome del cinque q meno”). Inoltre l’anemia refrattaria che può presentare delle alterazioni a carico dei globuli bianchi e delle piastrine è stata individuata come anemia refrattaria con displasia multilineare. Quindi la nuova classificazione WHO considera sempre l’anemia refrattaria, distinguendola sempre con sideroblasti e senza sideroblasti; la citopenia refrattaria con displasia multilineare (sempre con sideroblasti e senza sideroblasti) comprende quella che una volta era solo anemia refrattaria, però in questo caso è possibile considerare anche quei pazienti che hanno una neutropenia o una piastrinopenia in assenza di anemia, oppure considerare tutti quei pazienti che hanno oltre l’anemia anche la neutropenia e la piastrinopenia. Inoltre la classificazione WHO suddivide l'anemia refrattaria con eccesso di blasti in RAEB1(se la percentuale di blasti è inferiore al 10%) e RAEB2 (se la percentuale di blasti è superiore al 10%). Esistono comunque delle mielodisplasie non classificabili poiché non si riesce a definirle entro criteri diagnostici stabiliti. Questa classificazione ci permette di avere una migliore distribuzione in termini prognostici di queste patologie. Tuttavia per fare una valutazione prognostica non è sufficiente soltanto la classificazione WHO o la classificazione FAB, bisogna basarsi anche sull'IPSS (international prognostic scoring system). Tale sistema prende in considerazione tre parametri fondamentali che sono: 1. La percentuale di blasti 2. Le alterazioni del cariotipo 3. Il numero di citopenie A ciascuno dei parametri viene assegnato un punteggio. Se la percentuale di blasti è inferiore al 5% dà 0 come punteggio. Se la percentuale di blasti è tra il 5% e 10% viene assegnato 0.5 e se i blasti sono tra l'11% ed il 20% viene assegnato 1.5 come score. Le alterazioni del cariotipo possono essere favorevoli, intermedie o sfavorevoli. I cariotipi favorevoli sono: normale, mancanza del cromosoma Y, delezione del cromosoma 5 e del cromosoma 20. I cariotipi sfavorevoli sono quelli complessi e quelli caratterizzati dalla delezione del cromosoma 7 – esattamente come succede nelle leucemie acute. I cariotipi intermedi sono tutti gli altri. I cariotipi favorevoli, intermedi e complessi hanno score rispettivamente di 0, 0.5, 1 e infine al numero di citopenie (cioè a seconda che il soggetto sia soltanto anemico, piastrinopenico e neutropenico o più di una di queste citopenie), si dà un punteggio. Sommando i vari punteggi otteniamo un valore che ci permette di valutare la prognosi. I pazienti con score prognostico basso hanno una mediana di sopravvivenza di 5-6 anni; viceversa i pazienti con score alto (2 o più) hanno una sopravvivenza di soli quattro mesi. Quindi anche in questo caso, come spesso in molte malattie ematologiche neoplastiche, non conta l'etichetta della malattia sulla prognosi del paziente, ma è fondamentale individuare le caratteristiche della malattia nel singolo paziente che poi influenzeranno la prognosi. Quindi non vi limitate a classificare la malattia, ma ricercate i fattori importanti dal punto di vista prognostico. Questa valutazione prognostica è anche molto importante per definire il trattamento, considerando che si tratta di pazienti anziani: una terapia intensiva è poco indicata in gruppi a basso rischio, mentre una terapia intensiva è sicuramente indicata nel gruppo ad alto rischio. Nella normale ematopoiesi si ha il differenziamento delle tre linee maturative dalla cellula staminale, l'albero maturativo porta ad un numero normale di eritroiti, piastrine e leucociti. Nell'anemia refrattaria e nell'anemia refrattaria con sideroblasti invece la quota di elementi eritroidi è fortemente ridotta rispetto al normale e si ha pure una lieve riduzione per gli elementi della serie mieloide e piastrinica. Nell'anemia refrattaria con eccesso di blasti, accanto alle alterazioni che abbiamo visto, si verificano anomalie molto importanti a carico della linea mieloide, che rendono la malattia molto più vicina alle leucemie acute, si accumulano tutta una serie elementi blastici immaturi che non sono presenti invece nelle forme a basso rischio di mielodisplasia. La fisiopatologia delle mielodisplasie è molto complessa, sono tanti aspetti che intervengono nel cattivo funzionamento della cellula staminale tra cui: • aumento dell'apoptosi • alterazioni epigenetiche (alterazioni di espressione dei geni) • alterazioni del rapporto tra cellule stromali e cellule midollari • alterazioni del sistema immunitario • rilascio di citochine (IL1β e TNFα), determinanti un effetto inibitorio sulla crescita midollare • tossicità diretta che agisce direttamente sul microambiente midollare (chemioterapia e radioterapia). Queste cause possono determinare mielodisplasia, caratterizzata da alterazioni funzionali della cellula staminale e conseguenti modifiche numeriche e di funzione delle cellule normali. In questa bilancia tra proliferazione ed apoptosi dobbiamo considerare che nelle mielodisplasie a basso rischio c'è soprattutto un aumento dell'apoptosi; mentre in quelle ad alto rischio c'è una diminuzione del tasso apoptotico e contemporaneamente un aumento della proliferazione. Il quadro mielodisplastico passa generalmente da una condizione di emopoiesi inefficace (il midollo produce cellule che però vengono subito distrutte o vanno in apoptosi perché hanno dei difetti) ad un progressivo aumento di cellule francamente blastiche, che configurano la diagnosi finale di leucemia acuta mieloide. Quindi le mielodisplasie sono condizioni fortemente legate al rischio di evoluzione in leucemia mieloide acuta, alcune mielodisplasie (non tutte) possono essere considerate a tutti gli effetti condizioni preleucemiche dove il clone leucemico non si è ancora ben caratterizzato. Si passa quindi dalla condizione in cui c’è la mielodisplasia con un danno del DNA, un’accelerata apoptosi e una ematopoiesi inefficace a una condizione di leucemia acuta mieloide dove sicuramente si accumulano delle ulteriori alterazioni citogenetiche, con diminuzione dell'apoptosi, c’è una chiara trasformazione in leucemia. I quadri leucemici derivati da una mielodisplasia hanno una prognosi peggiore rispetto alla leucemia acuta de novo! Si tratta di leucemia molto gravi che di solito non rispondono al trattamento. In un modello di evoluzione di questa condizione possiamo ipotizzare una fase pre-MDS dove la cellula staminale subisce un evento tossico o anche per effetto di chemioterapia o di radiazioni (la chemioterapia essa stessa è fonte di evoluzione leucemica di una MDS, cioè può indurre prima la MDS e poi la trasformazione leucemica) o comunque ci possono essere delle alterazioni che non conosciamo molto bene (problemi di riparazione del DNA, enzimi) → tutto questo porta a una condizione di pre-mielodisplasia che poi diventa MDS con un danno diretto della cellula staminale che non riesce più a differenziare bene e che all’aumento della apoptosi associa una instabilità genomica che favorisce poi il passaggio alla fase finale di questa mielodisplasia dove come visto si riduce l’apoptosi ma aumenta la proliferazione e quindi c’è lo sviluppo di quella condizione di MDS in trasformazione leucemica → che evolve poi verso la leucemia acuta, la quale come detto è tra le peggiori leucemie acute in quanto è meno responsiva al trattamento. Una parte importante nella valutazione è svolta dalla citogenetica perché solo il 40% dei pazienti con mielodisplasia ha una citogenetica normale. Ricordiamo inoltre che la citogenetica è una valutazione abbastanza grossolana, non riesce ad evidenziare alcune microdelezioni, alcune microalterazioni e se andiamo a ricercare le alterazioni con metodiche più sofisticate, il valore di normalità si abbassa moltissimo! Fondamentalmente le mielodisplasie possono avere delle alterazioni citogenetiche a buona prognosi, cattiva prognosi e prognosi intermedia dove la buona prognosi è soprattutto rappresentata dal cariotipo normale e dal 5q- . Accanto ad alterazioni citogenetiche ci possono essere alterazioni epigenetiche. Il termine epigenetico indica una modificazione ereditaria (ereditaria non nel senso da padre a figlio ma da cellula a cellula) nel modello di espressione genica mediata da meccanismi differenti rispetto all'alterazione della sequenza nucleotidica primaria di un gene. Una mutazione epigenetica non è caratterizzata da sostituzione nucleotidiche o traslocazioni geniche ma è proprio l’espressione genica che viene modificata. In questo caso fondamentalmente si tratta di un'alterata metilazione del DNA; ci sono dei punti del DNA che sono più o meno metilati e che con il loro stato di metilazione influenzano la trascrizione e dunque l'espressione del gene. Nelle mielodisplasie si è dimostrata la presenza di ipermetilazione di alcuni geni oncosoppressori o di alcuni oncogeni che ne determinano una diversa espressione. Se noi abbiamo un gene oncosoppressore che viene ipermetilato, questo gene non si può esprimere ed in questo caso viene meno la sua funzione di controllo della proliferazione cellulare. Viceversa in alcuni casi gli oncogeni possono avere alterati livelli di metilazione del DNA con conseguente alterata espressione, che può portare a trasformazione neoplastica. Le mielodisplasie sono malattie dell’età anziana, la mediana di comparsa della malattia è intorno ai 70 anni e usualmente sono paucisintomatiche e di scarsa obiettività clinica, in rari casi il paziente è splenomegalico, ma nella maggior parte dei casi non troviamo nulla all'esame fisico. Nella maggior parte dei casi il paziente va dal medico perché si sente stanco, la stanchezza è dovuta all'anemia, mentre la piastrinopenia e la leucopenia, essendo di modesta entità, non causano problemi al paziente. A questo punto si fa l'emocromo e si vede l'alterazione della normale emopoiesi, si può riscontrare infatti una citopenia che può essere pan-, bi- o monolineare. E’ molto importante il fatto che questi pazienti, per questa condizione ma anche per il fatto che c’è una disregolazione del sistema immunitario spesso presentano delle infezioni, soprattutto batteriche. In qualche caso, quando c’è una grave piastrinopenia, ci può essere un sanguinamento. Le cause di morte del paziente mielodisplastico sono soprattutto le infezioni e le trasformazioni in leucemie mieloidi acute. Ci sono poi le cosiddette comorbidità perché i pazienti sono spesso anziani e quindi la morte può avvenire per malattie cardiovascolari o altre patologie tipiche dell'anzianità. Diagnosi La diagnosi si fa con criteri morfologici (displasie a livello midollare), dimostrando la citopenia periferica pan-, bi- o monolineare con l'emocromo e le alterazioni citogenetiche che sono presenti nel 40-70% dei pazienti affetti da questa malattia. E’ chiaro che bisogna porre attenzione alla diagnosi differenziale con altre patologie, molto spesso questi pazienti hanno un'anemia macrocitica e dunque la diagnosi differenziale più frequente è con l'anemia da carenza di B12 o di acido folico, che rappresentano le condizioni più frequenti di anemia macrocitica. Anche il piombo può dare una citopenia periferica che deve essere distinta dalle condizioni mielodiplastiche. Alcune esposizioni ad antibiotici, alcune infezioni virali, le malattie infiammatorie (ricordiamo che nelle malattie infiammatorie esiste la cosiddetta anemia da flogosi cronica dovuta all'eccessiva produzione di epcidina che impedisce la corretta mobilizzazione del ferro, con aumento dei valori di ferritina che spesso ritroviamo anche nelle mielodisplasie), l'anemia aplastica e la mielofibrosi devono essere tutte valutate nella diagnosi differenziale. Qual è la possibilità di evoluzione di una mielodisplasia? Nell'evoluzione verso un quadro di leucemia acuta mieloide possiamo avere pazienti con un decorso cronico protratto; pazienti con decorso cronico un po’ più aggressivo caratterizzato dal fatto che solo dopo molti anni evolve in leucemia acuta mieloide. Si può riscontrare un decorso subacuto, in cui dopo un periodo relativamente breve si avrà l'evoluzione verso la leucemia. Possiamo infine avere un decorso acuto caratterizzato dalla rapida evoluzione verso la leucemia acuta mieloied. Terapia Per fare un trattamento corretto bisogna prima di tutto distinguere tra malattie a basso rischio ed ad alto rischio. Nelle malattie a basso rischio dobbiamo cercare di migliorare i parametri ematologici e quindi una terapia un po’ più palliativa, tenendo conto molto della qualità di vita del paziente. Ricordiamo che molto spesso si tratta di pazienti anziani con prospettiva di vita di cinque o sei anni al massimo, quindi non bisogna aggredirli con pesanti cicli di chemioterapia ma dobbiamo ricercare una terapia di supporto cercando di migliorare la loro qualità di vita. Viceversa in pazienti con alto rischio di trasformazione neoplastica, con alto rischio di morire per la malattia, anche se si tratta di pazienti anziani, bisogna cercare in qualche modo di alterare la storia di questa malattia con terapie più aggressive, considerando però sempre la qualità di vita del paziente. Il caposaldo della terapia delle mielodisplasie rimane la trasfusione di sangue. Si possono trasfondere i soli globuli rossi o le sole piastrine nei pazienti piastrinopenici. Bisogna però considerare che le trasfusioni multiple inducono delle complicazioni, inducono sensibilizzazione soprattutto per quanto riguarda le piastrine ma anche i globuli rossi; si può avere un sovraccarico di ferro, che dovrà essere chelato per evitare una emocromatosi secondaria. Per evitare queste complicazioni recentemente è stata proposta la terapia con eritropoietina. L'industria farmaceutica oggi ci mette a disposizione l'eritropoietina ricombinante, un farmaco che spesso somministriamo ai pazienti mielodisplastici. Tuttavia non sempre i pazienti rispondono alla somministrazione di eritropoietina, grossomodo risponde solo il 30% dei pazienti e quelli che rispondono sono i pazienti che non hanno alterazioni citogenetiche o non hanno avuto bisogno di una precedente trasfusione, sono pazienti che presentano bassi livelli di eritropoietina endogena e che solitamente non hanno sideroblasti ad anello: in definitiva se consideriamo questi parametri possiamo concludere che l'eritropoietina funziona purtroppo soltanto nei pazienti migliori ma non in quelli peggiori, ovvero quei pazienti che fanno molte trasfusioni, che hanno un elevato dosaggio endogeno di eritropoietina e pazienti che hanno delle anomalie citogenetiche. Pertanto in questi pazienti ad alto rischio oggi si cerca di intervenire sulle alterazioni epigenetiche che dicevamo prima. Sono farmaci che interferiscono sulla metilazione e sulla deacetilazione del DNA. Ci sono alcuni farmaci come l'azacitidina, la decitabina o l'acido valproico che sono in fase avanzata di sperimentazione o sono già approvati e applicati in trials clinici. Uno dei trial più importanti è quello che ha messo a confronto la terapia con azacitidina con la sola terapia di supporto, in pazienti con mielodisplasia ad alto rischio. Si può vedere che i pazienti trattati con azacitidina vanno meglio rispetto a quelli trattati con la sola terapia di supporto. Tuttavia i dati non sono pienamente soddisfacenti perché non si raggiunge un plateau ma solo un ritardo nella trasformazione leucemica. Tuttavia questi sono piccoli passi che sono necessari per ottenere un miglioramento nella prognosi di questi pazienti. Tuttavia rimane purtroppo evidente che le attuali terapie incidono in maniera blanda sulla storia naturale di queste malattie. · Un altro farmaco che sta avendo dei successi importanti è la Lenalidomide (derivato della Talidomide), se ricordate è lo stesso farmaco che abbiamo incontrato parlando del mieloma, è un farmaco che si è dimostrato particolarmente efficace nel trattare un sottotipo di mielodisplasia che è la cosiddetta Sindrome del 5q - . La Lenalidomide è un farmaco particolare che agisce: 1 2 3 4 riducendo i livelli di TNF (aumentati nelle mielodisplasie) riducendo la neoangiogenesi aumentando la risposta immunologica (spesso alterata nelle mielodisplasie) aumentando il signalling del recettore per l'eritropoietina (aumenta così l'attività dell'eritrone). La Lenalidomide quindi si è dimostrata particolarmente utile in questo sottogruppo di pazienti con la sindrome del 5q -. La sindrome del 5q- è detta appunto sindrome perché è una condizione dove non c’è soltanto una delezione cromosomica ma è caratterizzata anche da alcuni aspetti peculiari: colpisce il sesso femminile; è caratterizzata da anemia macrocitica con modesta leucopenia e le piastrine addirittura sono spesso aumentate, non diminuite. Inoltre a livello midollare è caratteristica la presenza di megacariociti ipolobulati. In genere è una condizione a basso rischio di trasformazione leucemica che avviene soltanto in un 10% dei pazienti. E’ vero che non tutte le sindrome del 5q- sono uguali, nel senso che la sola delezione del 5q è una situazione abbastanza rara (5% di tutte le mielodisplasie), è più frequente l'associazione tra la delezione del 5q ed altre mutazioni. In ogni caso si tratta di sindromi piuttosto peculiari con questa anemia macrocitica e modesta leucopenia, con le alterazioni tipiche dei megacariociti, con un decorso indolente e sono particolarmente sensibile alla Lenalidomide, il trattamento della sindrome del 5q- induce una risposta completa nell'80% dei pazienti. Ricordiamo che la classificazione di queste malattie è molto complessa: c’è una sovrapposizione tra il gruppo delle leucemie acute mieloidi e il gruppo delle mielodisplasie, alcune malattie sono a cavallo tra leucemie mieloidi acute e le mielodisplasie. Come anche la mielodisplasia è a cavallo con tutta una serie di altre malattie, soprattutto la cosiddetta aplasia midollare, che gli autori anglosassoni chiamano anemia aplastica. * Se ricordiamo l'anemia aplastica è un disturbo che si associa all'emoglobinuria parossistica notturna e quindi c'è un forte overlapping tra queste due patologie. Anche l'aplasia eritroide pura e tante malattie midollari complicano notevolmente la classificazione. Questo a dimostrare come non sempre è facile fare una classificazione di queste malattie e le mielodisplasie ipocellulari vanno sicuramente in diagnosi differenziale con l’aplasia midollare (o anemia aplastica). L'anemia aplastica è una condizione, per fortuna rara, che può essere dovuta a: 1. Farmaci citotossici o radiazioni 2. Alcune condizioni tossiche per il midollo, primo fra tutti il benzene 3. Alcuni virus come il virus dell’epatite B Il virus dell'epatite B è frequentemente associato all'aplasia midollare. Si tratta di condizioni estremamente gravi, quasi tutti i pazienti con aplasia midollare secondaria ad epatite B non rispondono al trattamento. In questa biopsia ossea potete notare come si notino le trabecole ossee, però con la totale assenza di midollo nelle trabecole. Per questi pazienti si può avviare una terapia immunosoppressiva (nei pazienti anziani) o si può eseguire il trapianto di midollo (nei pazienti giovani). Il trattamento dei pazienti con immunosoppressori è giustificato dal fatto che si suppone un importante ruolo del sistema immunitario nella patogenesi dell'aplasia midollare. Esiste pure la cosiddetta aplasia eritroide pura, caratterizzata dalla sola deplezione dell'eritrone, cioè scompare la produzione di globuli rossi e questa condizione è spesso associata al timoma. Quando c'è un paziente con timoma andate a guardare l'emocromo perché si potrebbe configurare un quadro di deplezione della linea eritroide e quindi l’aplasia eritroide pura. LEUCEMIA MIELOIDE CRONICA Definizione La Leucemia Mieloide Cronica è una malattia mieloproliferativa caratterizzata da aumentata produzione di granulociti, senza perdita della capacità di differenziazione (quindi differente rispetto alla leucemia acuta mieloide) e presenza di mieloproliferazione a livello splenico ed epatico. La LMC rappresenta il 20% di tutte le leucemie dell’età adulta, compare in forma cronica e stabile e progredisce verso una forma acuta dopo un periodo di tempo variabile, quindi è una malattia cronica che può progredire verso una forma acuta. E’ una malattia abbastanza rara, cioè soltanto 1-2 casi per anno per ogni 100 mila abitanti, ad es. in una città come Catania che fa mezzo milione di abitanti, se ne vedono da 5 a 10 casi l’anno. (slide) Il quadro ematologico è un quadro tipico, allo striscio periferico di un paziente con LMC si riconoscono tanti granulociti e tanti elementi più immaturi della serie mieloide (meta mielociti, pro mielociti), cioè tutto lo spettro della linea di maturazione mieloide, dal mieloblasto, al pro mielocita, al meta mielocita e infine al granulocita. Patogenesi Questa malattia è caratterizzata in maniera patognomonica da una alterazione cromosomica rappresentata dal famoso cromosoma Philadelphia, così chiamato proprio dalla città dove è stato descritto per la prima volta. Il cromosoma Philadelphia, che abbiamo visto anche nelle leucemie acute, è in realtà tipico della LMC ed è dato dalla traslocazione fra il cromosoma 9 e il cromosoma 22. Questa traslocazione provoca un trascritto ibrido che prende il nome di bcr-abl, il quale porta poi ad una proteina ibrida che prende il nome di proteina ibrida BCR-ABL → questa proteina presenta attività tirosin-chinasica costitutiva. E’ stato dimostrato con esperimenti fatti nel topo di laboratorio che se prendiamo un topo e inseriamo il gene bcr-abl, questo sviluppa una malattia che è molto simile alla LMC, si è avuta così la prova che questa traslocazione è l’evento responsabile della trasformazione leucemica della cellula. Quindi il cromosoma Philadelphia è il marcatore caratteristico della Leucemia Mieloide Cronica ed è presente potremmo dire nel 100% dei casi: anche se magari in citogenetica a volte non si vede questa traslocazione, se andiamo a verificare in biologia molecolare, cioè andiamo a ricercare il trascritto molecolare per il bcr-abl lo ritroviamo nel 100% dei casi! Il cromosoma Philadelphia si ritrova anche in: - circa il 30% delle leucemie acute linfoblasti che dell’adulto - il 2% delle leucemie acute mieloidi Il gene abl dal cromosoma 9 passa nel cromosoma 22 e qui forma il trascritto ibrido bcr-abl. - Il bcr è un gene di espressione ubiquitaria che ha una localizzazione citoplasmatica, ma durante la mitosi si trova in sede pericromosomica, e nei topi mancanti del gene (topi knock-out) c’è un’emopoiesi assolutamente normale, quindi la funzione di questo gene non è ben chiarita. - Per quanto riguarda il gene abl i topi knock-out per questo gene sono leucopenici e la proteina ABL (che probabilmente è quella veramente importante) ha un’attività tirosin-chinasica e determina proliferazione cellulare e inibizione dell’apoptosi. Quando c’è la traslocazione ovviamente si rompono sia l’abl che il bcr e la parte di abl che dal 9 migra sul 22 è sempre la stessa mentre invece il punto di rottura del gene bcr può essere differente, tra l’altro bcr significa proprio break-point cluster region. A seconda del punto di rottura si possono distinguere una condizione di major breakpoint-clusterregion (M-bcr), minor bcr (m-bcr) e il μ-bcr, quest’ultima molto rara (pronuncia “miù bcr”). Pertanto a seconda del punto di rottura il gene di fusione che si viene a creare bcr-abl ha una lunghezza differente e questo come visto dipende dal bcr perché l’abl ha sempre la stessa lunghezza. Quindi ad esempio se il bcr si è rotto nel punto di major breakpoint-cluster-region (M-bcr) avremo una proteina di 210 dalton che è tipica della leucemia mieloide cronica. Se invece si è avuta una rottura del bcr nel m-bcr avremo una proteina con un peso molecolare di 190 e questa è presente soltanto nella leucemia acuta linfoblastica (LLA). Infine il μ-bcr si trova soltanto in quei pazienti che hanno una cosiddetta leucemia neutrofila cronica (LCN), ovvero una condizione in cui nel sangue periferico troviamo quasi esclusivamente neutrofili e non troviamo gli elementi più immaturi della serie mieloide (ma questa ripetiamo è una condizione piuttosto rara). Quindi le caratteristiche della proteina BCR-ABL quali sono? Contrariamente alla sua controparte normale (proteina ABL), questa proteina BCR-ABL è presente soltanto nel citoplasma e non nel nucleo ed è continuamente in uno stato di attivazione, insensibile ai normali meccanismi di controllo, è in grado di fosforilare numerosi bersagli e di conseguenza induce un’attivazione continua di alcune sub unità cellulari: l’eccesso di attività di questa proteina è quello che poi determina la trasformazione neoplastica della cellula mieloide. Per far questo è necessario che il BCR-ABL dimerizzi, quindi due molecole di BCR-ABL che si fondono insieme e che poi determinano un’attivazione costitutiva della tirosin-chinasi, questa va a fosforilare numerosi substrati che servono alla proliferazione della cellula. Quindi fondamentalmente questa proteina anomala BCR-ABL che si trova nel citoplasma, determina: - la fosforilazione di una serie di substrati a valle come JAK-STAT, MYC che servono come stimolo alla proliferazione - determina anche un’alterazione di alcune proteine (paxillin) che servono all’adesione di queste cellule alla matrice midollare → di conseguenza queste cellule non hanno più la capacità di aderire allo stroma midollare e passano nel sangue periferico! Ecco perché noi in questi pazienti vediamo una leucocitosi, che non è soltanto espressione della proliferazione del clone neoplastico ma è anche espressione del fatto che il clone neoplastico ha perso l’ancoraggio alla matrice extracellulare del midollo osseo e quindi passa anche nel sangue periferico. - viene inibita l’apoptosi e quindi da un lato lo stimolo proliferativo, dall’altro l’inibizione dell’apoptosi, si capisce bene che queste cellule aumentano di molto (ad es. l’altro giorno il prof. ha visto un paziente che aveva 600 mila globuli bianchi!!) - infine una conseguenza molto importante è quella della diminuzione della funzione di ABL a livello del nucleo che provoca una instabilità genomica, significa che queste cellule possono accumulare mutazioni genomiche che le rendono sempre più neoplastiche, sempre più maligne. L’instabilità genomica fa sì che queste cellule Philadelphia positive possano nel tempo accumulare dei difetti genetici addizionali per cui non solo si sostituiscono alle cellule normali, man mano diventano anche più maligne → pertanto alla fine queste cellule diventano cellule con un clone molto indifferenziato che configura la cosiddetta crisi blastica. Questa ricordiamo è una malattia che nella sua storia naturale nasce come malattia cronica ma poi evolve invariabilmente verso una leucemia acuta e si tratta di una leucemia acuta di quelle assolutamente resistenti al trattamento perché sono cellule che hanno acquisito tante di quelle alterazioni genetiche, genomiche che non sono più sensibili ad alcun trattamento. Diagnosi La diagnosi si fa sulla base delle caratteristiche che abbiamo visto in termini morfologici ma soprattutto per fare la diagnosi occorre documentare la presenza del cromosoma Philadelphia (diagnosi citogenetica) o del suo trascritto molecolare, cioè del gene di fusione bcr-abl. La diagnosi differenziale va posta con la cosiddetta reazione leucemoide, una condizione in cui a causa di infezioni, tumori, stati infiammatori c’è una condizione che stimola la proliferazione di elementi midollari e quindi stimola la produzione di neutrofili e anche di meta mielociti che possono passare nel sangue periferico, così vediamo pazienti che hanno 20-30 mila globuli bianchi con un aumento del numero di cellule immature nel sangue periferico, ma queste sono delle condizioni secondarie a delle stimolazioni midollari (questi pazienti ovviamente non hanno il cromosoma Philadelphia). Bisogna distinguere da altre malattie mieloproliferative come le policitemia vera e la trombocitemia essenziale che possono stimolare anche il clone mieloide ad aumentare. Inoltre ci può essere una negatività del gene di fusione perché il gene è coinvolto in altre traslocazione ma questa situazione è abbastanza rara. La maggior parte dei pazienti sono asintomatici, cioè vengono dall’ematologo perché per caso hanno fatto un emocromo e si vede che hanno una spiccata leucocitosi, in questi pazienti quindi la diagnosi si fa per caso. Alla visita del paziente spesso troviamo una splenomegalia e se chiediamo il paziente ci dirà che da qualche mese notava una certa astenia; alcuni pazienti presentano anche sintomi più spiccati quali la febbricola e altri sintomi generali ma fondamentalmente quello che ci fa fare la diagnosi è la presenza nell’emocromo di questa spiccata leucocitosi con presenza nel sangue periferico di una granuloblastosi, cioè di un aumento non solo dei granulociti neutrofili ma anche dei mielociti, pro mielociti etc. A volte quando c’è un eccesso di leucocitosi (in alcuni pazienti si arriva anche a 600 mila globuli bianchi), in quel caso ci può essere anche un problema di viscosità con disturbi della vista, sordità, vertigini, problemi cardiovascolari. A volte non solo aumentano i globuli bianchi ma aumentano anche le piastrine, non sono rari i pazienti con LMC in cui c’è un aumento significativo delle piastrine (possiamo arrivare anche a 1 milione e mezzo di piastrine) e generalmente queste condizioni si possono accompagnare anche a trombosi sia centrali (TIA, disturbi visivi) sia periferiche con trombosi delle dita o delle vene profonde. Tuttavia in condizioni rare un eccesso di piastrine può provocare anche un’emorragia, può sembrare paradossale che un aumento delle piastrine provochi emorragia: probabilmente questo dipende dal fatto che questo eccesso di piastrine consuma il fattore di Von Willebrand (fattore che serve a far aderire le piastrine al sub-endotelio), per cui quando ci sono troppe piastrine viene consumato, è come se il paziente avesse un difetto di questo fattore della coagulazione. Quindi all’esame emocromocitometrico: - quantità normale o lievemente ridotta di Hb - le piastrine sono più spesso normali oppure aumentate - i leucociti sono costantemente aumentati e vanno da 20 mila a 500 mila Il quadro midollare è piuttosto caratteristico e si configura quello che viene tecnicamente chiamato midollo packed, “impacchettato”, pieno di elementi cellulari Decorso e Prognosi Quasi tutti i pazienti nascono in fase cronica però invariabilmente, se questa malattia non viene trattata, tutti i pazienti evolvono in una fase blastica, alcuni evolvono in maniera rapida e diretta altri con una fase intermedia che viene detta fase accelerata. E’ chiaro che su questa trasformazione intervengono numerosi fattori genetici e molecolari, ad esempio ci può essere l’inattivazione di p53, accorciamento dei telomeri ma fondamentalmente tutto dipende da quella instabilità genomica che hanno queste cellule nella fase cronica: tutte queste condizioni poi trasformano la leucemia mieloide cronica da una fase cronica (paucisintomatica o asintomatica, il paziente sta bene e presenta soltanto una iperleucocitosi) verso una malattia assolutamente mortale in pochi mesi, cioè verso una leucemia acuta del tutto refrattaria che prende il nome di crisi blastica. La fase cronica, stabile dura da 1 a 5 anni, caratterizzata da anemia moderata, incremento dei globuli bianchi, splenomegalia → successivamente si passa ad una fase accelerata o cosiddetta di trasformazione che è caratterizzata sicuramente da un aumento dei blasti, un peggioramento dell’anemia oppure da una piastrinopenia → infine la cosiddetta crisi blastica che si identifica come tale quando ci sono più del 30% di blasti nel midollo ed è usualmente una crisi blastica mieloide (ma non vi stupite di trovare in alcuni casi una crisi blastica linfoide!! cioè passiamo da una patologia che è definita Leucemia Mieloide Cronica a una crisi blastica che è di tipo Linfoide). Questo avviene fondamentalmente perché si tratta di una malattia della cellula staminale, che inizialmente si estrinseca, si esprime con una proliferazione del clone mieloide ma quando appunto le alterazioni citogenetiche, molecolari sono tali da indirizzare la cellula staminale verso il differenziamento linfoide allora in questo caso la crisi blastica può essere anche Linfoide. La crisi blastica quindi può essere sia mieloide che linfoide e a volte è anche mista, ci sono delle cellule che hanno una caratteristica sia di cellule mieloidi che di cellule linfoidi: in questo caso si parla di crisi blastica bifenotipica. Anche per la Leucemia Mieloide Cronica si possono individuare dei fattori di prognosi, in particolare ci sono 2 classificazioni più importanti: Sokal Score – basato sull’età, la splenomegalia, la percentuale di blasti nel sangue periferico e sul numero di piastrine, poi c’è una formula matematica molto complicata che si fa al computer e che ci dà uno score prognostico Hasford Score – anche questo basato sull’età, la splenomegali, la percentuale di blasti nel sangue periferico, piastrine ma questo score prende in considerazione anche la percentuale di basofili e di eosinofili nel sangue periferico che hanno un valore prognostico negativo, cioè se in questi pazienti troviamo molti basofili e molti eosinofili, non sappiamo bene per quale motivo, ma sono pazienti che evolvono più rapidamente nella fase blastica. Terapia Per la terapia della LMC bisogna considerare che di recente sono stati definiti dei criteri di risposta che in precedenza non erano identificati, prima infatti era una malattia assolutamente non guaribile, una malattia sulla quale potevano intervenire poco. Con il passare del tempo si è passati da una terapia palliativa rappresentata da busulfano, idrossiurea a una terapia con intenti curativi rappresentata prevalentemente negli ultimi anni dal trapianto di midollo, che è stato preceduto o comunque integrato dalla terapia con IFN o dalla chemioterapia. La terapia di recente ha subito un’accelerazione improvvisa per l’impiego in clinica di farmaci che hanno un’attività specifica contro il cromosoma Philadelphia che sono l’Imatinib - il famoso Glivec, un farmaco entrato in commercio in Italia nel 2003 che ormai ha guadagnato pure le prime pagine dei quotidiani - e i farmaci di II generazione che sono il Dasatinib e il Nilotinib. Tornando al concetto iniziale, proprio per la disponibilità di farmaci che producono queste risposte, è stato possibile iniziare a definire le risposte. Quindi adesso noi consideriamo: - la risposta ematologica completa, cioè con la normalizzazione dei valori dell’esame emocromo e assenza di milza o fegato palpabile, quindi il paziente che ha una risposta ematologica completa ha una normalizzazione dei parametri ematologici: tuttavia non ci possiamo accontentare di questo, tali risultati infatti li ottenevamo anche con la chemioterapia convenzionale, dobbiamo ottenere invece la risposta citogenetica. - si definisce risposta citogenetica maggiore cioè quando ci sono meno del 33% di cellule Philadelphia positive - si parla di risposta citogenetica completa quando scompaiono completamente le cellule Philadelphia positive - ma accanto a questi c’è anche la risposta molecolare, perché noi andiamo a valutare non soltanto la citogenetica ma anche il trascritto molecolare (il famoso bcr-abl) e in questo caso consideriamo una risposta molecolare maggiore quando il rapporto bcr-abl / abl è inferiore a 0.1. - si considera infine risposta molecolare completa quando anche con metodiche di biologia molecolare non riusciamo a evidenziare alcun trascritto molecolare bcr-abl. Si può osservare come si tratta di una valutazione progressiva, sempre più profonda, perché il limite di detection della risposta ematologica completa è fra 1 e 5%; il limite di detection della risposta citogenetica completa è dell’1% ma il limite di detection della risposta molecolare è di 1-5, cioè di 1/100000, quindi si capisce bene come andiamo a valutare livelli di risposta in maniera sempre più profonda e sempre più precisa. La chemioterapia convenzionale è basata sull’idrossiurea e il busulfano, ma questo ha un effetto soltanto palliativo e si utilizza in pazienti molto anziani. In passato sono stati utilizzati anche l’Interferone α e la citosina arabinoside, anche in questo caso, nonostante l’IFN sia stato il primo farmaco a intaccare l’alterazione citogenetica, cioè il primo farmaco in grado di ridurre la percentuale di cellule Philadelphia positive, anche qui la guarigione non era assolutamente alla nostra portata. L’intervento che invece faceva guarire i pazienti era invece il trapianto di midollo allogenico che faceva guarire circa il 60% dei pazienti in fase cronica, però doveva essere un paziente giovane (età < a 45 anni) e con un donatore HLA compatibile e la mortalità del trapianto è di circa il 20%, per non considerare la possibilità di recidive. Quindi in caso di trapianto con un paziente di età > 45 anni in questo caso il rischio di mortalità è molto elevato ed è aumentato anche qualora il donatore non sia consanguineo; inoltre il trapianto andava fatto in fase cronica, se veniva fatto in fase accelerata o blastica non era più efficace. · Per fortuna nel 2001 (in Italia dal 2003) abbiamo a disposizione l’Imatinib, un farmaco studiato in maniera specifica per andarsi a sostituire all’ATP con cui agisce la proteina BCR-ABL, per cui l’Imatinib si inserisce nella tasca al posto dell’ATP. In questo modo la proteina BCR-ABL (tirosin-chinasi) non può più utilizzare l’ATP, non può più fosforilare il suo substrato e quindi non può più esercitare la sua azione di tirosin-chinasi. Pertanto l’Imatinib agisce in maniera specifica soltanto sulle cellule che hanno questa anomalia (bcr-abl) e testato su linee cellulari non è in grado di indurre una citolisi di linee cellulari che sono bcr-abl negative mentre è in grado di indurre citolisi sulle cellule bcr-abl positive. Lo studio che rimane più importante è stato il cosiddetto studio Iris che metteva a confronto l’ Imatinib con quella che era definita fino al 2001 la terapia migliore ovvero l’IFN + Ara-C. Lo studio Iris ha consacrato la terapia con Imatinib come quella che induce la maggiore percentuale di risposte ematologiche complete ma soprattutto la maggiore percentuale di risposte citogenetiche complete: solo il 14% dei pazienti trattati con IFN-Ara-C ottenevano la risposta citogenetica completa contro il 76% dei pazienti trattati con Imatinib! Tra l’altro l’IFN è molto meno tollerato rispetto all’ Imatinib. La curva di sopravvivenza dei pazienti trattati con Imatinib è molto vicina al 100%, è stato calcolato che adesso i pazienti con Leucemia Mieloide Cronica hanno una curva di sopravvivenza che è quasi sovrapponibile a quella dei pazienti diabetici di uguale età e sesso, se consideriamo che la LMC era una malattia inevitabilmente mortale fino al 2001, c’è adesso una gran bella differenza! Quando andiamo a valutare il paziente trattato con Imatinib, lo andiamo a valutare non più ovviamente soltanto con l’emocromo e con la visita (quello è implicito) bensì con la citogenetica ma soprattutto con la biologia molecolare poiché quello che dobbiamo ottenere, l’obiettivo del nostro trattamento è la remissione molecolare maggiore, cioè come visto meno dello 0.1% nel rapporto bcr-abl / abl. Perché in effetti se andiamo a valutare meglio la curva osserviamo che i pazienti che non hanno ottenuto la remissione citogenetica completa sono i pazienti che vanno sicuramente peggio, ma anche i pazienti che hanno ottenuto la remissione citogenetica completa ma non hanno una remissione molecolare maggiore (in termini di riduzione del trascritto molecolare), in effetti anche questi vanno un po’ peggio rispetto a chi la ha ottenuta. In questi pazienti quindi occorre misurare ogni 3 mesi il trascritto molecolare attraverso metodiche di biologia molecolare poiché questo ci consente di capire se il paziente continua a mantenersi in remissione. Effetti collaterali L’ Imatinib però non è del tutto privo di effetti collaterali, presenta una certa tossicità ematologica anche se abbastanza modesta e non preoccupante tuttavia l’effetto collaterale più importante è l’edema, sono pazienti che presentano degli edemi usualmente abbastanza diffusi, ad es. l’edema periorbitale è una costante nei pazienti che fanno l’ Imatinib (soltanto nel 3% dei casi si tratta di un edema grave). Bisogna anche dire che non tutti i pazienti rispondono all’ Imatinib, alcuni sviluppano delle resistenze, delle intolleranze o semplicemente i pazienti stanno bene e non prendono il farmaco, oppure prendono dei farmaci che interferiscono con l’ Imatinib. Vi sono tutta una serie di possibilità che determinano nel paziente una risposta minore al farmaco ma di fronte a tutte queste varabili ci sono anche i casi di vere e proprie resistenze al farmaco, che possono dipendere da un’evoluzione clonale della malattia, cioè ci sono alcune alterazioni della cellula, del clone neoplastico che rendono questa cellula non più sensibile all’ Imatinib. Per questo sono stati studiati degli altri farmaci, il Nilotinib in particolare (che può essere considerato una specie di super Imatinib) e il Dasatinib, farmaci in grado di revertire la resistenza in quei pochi pazienti che sono resistenti all’ Imatinib. Purtroppo comunque se il paziente va in crisi blastica non c’è niente da fare, anche se trattati con dosaggi alti, questo dipende dal fatto che l’ Imatinib è in grado di eliminare tutte le cellule più mature ma non è in grado di eliminare le cellule staminali. La sfida per il futuro è quella di trovare farmaci simili all’ Imatinib ma che siano in grado di eliminare anche le cellule staminali, al momento noi siamo in grado di ottenere spettacolari curve di sopravvivenza ma non riusciamo a far guarire i pazienti, i pazienti sono costretti ad assumere quotidianamente 4 pillole di Imatinib per tutta la vita perché altrimenti vanno in progressione. Lezione 14 Test sulle mielodisplasie: • Le sindromi mielodisplastiche sono caratterizzate spesso da una monocitopenia, bi-citopenia o una pancitopenia • I soggetti più colpiti dalla sindrome mielodisplastica sono gli uomini anziani • I fattori di rischio più importanti sono rappresentati sia dalla percentuale di blasti, sia la citogenetica che l’entità della citopenia (ovvero se è solo una mono-citopenia oppure bi- o pan citopenia): queste tre condizioni poi configurano quello che viene definito IPSS (International Prognostic Scoring System) che attribuisce un punteggio differente a ognuna di queste variabili • La sindrome del 5q – è una sindrome peculiare che non identifica soltanto l’alterazione citogenetica (delezione del cromosoma 5), ma identifica una vera e propria sindrome con tutta una serie di alterazioni caratterizzate appunto da megacariociti ipolobulati, piastrine aumentate (condizione questa che raramente si riscontra nelle altre sindromi mielodisplastiche dove invece c’è di frequente piastrinopenia). Ha una prognosi favorevole ed è prevalente nel sesso maschile. • Il sovraccarico di ferro ha come bersaglio soprattutto cuore, fegato e ghiandole endocrine. Anche i reni possono essere bersaglio ma questo non ha un’importanza clinica così come ce l’hanno a carico del fegato, del cuore e delle gh. endocrine • La terapia con EPO ricombinante purtroppo è efficace soltanto nei pazienti con buona prognosi, con bassa concentrazione endogena di EPO. • I pazienti con anemia aplastica possono presentare anemia- leucopenia- piastrinopenia. Il prof ha messo questa domanda perché non vuole che si generasse l’equivoco e la sbagliata convinzione che l’anemia aplastica interessa solo i globuli rossi, infatti soprattutto gli Autori americani parlano di anemia aplastica per indicare una condizione di aplasia midollare in cui c’è un difetto di tutte e tre le linee. • L’aplasia midollare può essere conseguenza di molte situazioni ma quella più frequente e più grave è quella del virus dell’epatite B. Test sulla leucemia mieloide cronica: • I fattori di prognosi più importanti nella leucemia mieloide cronica sono la basofilia, la leucocitosi e la splenomegalia mentre invece l’età non ha molta importanza nella leucemia mieloide cronica. • La crisi blastica è una malattia che coinvolge la cellula staminale ematopoietica, per cui se nella prima fase si manifesta con una proliferazione mieloide, in una fase avanzata, cioè durante la crisi blastica, è la stessa cellula staminale che può differenziarsi verso la linea mieloide o linfoide o addirittura dare luogo ad una popolazione con fenotipo misto, si parla di bifenotipo. • Il cromosoma Philadelphia che è il marcatore diagnostico della leucemia mieloide cronica, può essere presente anche nelle leucemie acute linfoidi, dove configura un fattore prognostico negativo (può essere presente anche nelle Leucemie acute mieloidi, ma è molto raro). Per esempio una delle caratteristiche differenziali tra la leucemia acuta Linfoide del bambino e quella dell’adulto, è rappresentata dal fatto che nel bambino il cromosoma Ph è molto raro, mentre nell’adulto è molto frequente. • La chemioterapia induce una risposta citogenetica soltanto nel 5% dei pazienti, questo a dimostrazione del fatto che la chemioterapia non è una terapia efficace nella LMC • Una volta si usava nella terapia l’INF alfa, questo è un farmaco che ha tanti effetti collaterali tra cui il più importante è la cosiddetta “flu like sindrome”, i sintomi dell’influenza (malessere - mal di testa – dolori ossei) sono dovuti al fatto che il nostro organismo produce citochine in risposta al virus e una di queste è l’INF. Quindi è chiaro che se andiamo a somministrare IFN in questi pazienti, avremo tutti gli effetti di una sindrome influenzale alla quale però poi usualmente i pazienti si abituano, dopo 1-2 settimane dall’inizio della somministrazione. • L’STI è l’inibitore della tirosin-chinasi il “glirech” ALTRE MALATTIE MIELOPROLIFERATIVE CRONICHE Oggi completiamo le mieloproliferative croniche. In una rappresentazione schematica delle patologie ematologiche neoplastiche distinguiamo: 1. Linfoidi 2. Mieloidi: acute e croniche Tra le forme croniche abbiamo già parlato della Leucemia Mieloide Cronica, rimangono da trattare le altre malattie mieloprofliferative croniche che sono fondamentalmente: - la Policitemia Vera - la Trombocitemia essenziale - la Mielofibrosi con metaplasia mieloide (slide) questo è un quadro ematologico di policitemia vera: rispetto al sangue normale, nel soggetto policitemico l’ematocrito è molto maggiore a causa di un aumento significativo dei GR e quindi percentualmente si riduce la parte plasmatica e aumenta la parte corpuscolata. Il midollo di questo paziente è ipercitosico, aumenta la quota di elementi eritroidi e c’è un lieve aumento della quota retico litica cioè della fibrosi midollare. (slide) Questo è un quadro di trombocitemia essenziale: nell’ematocrito abbiamo un aumento delle piastrine, nel midollo vi è un aumento dei megacariociti. (slide) La mielofibrosi idiopatica infine è una condizione in cui c’è una notevole anisopoichilocitosi dei GR e le emazie a lacrima (tear-drop shaped), che sono tipiche della mielofibrosi, a livello del midollo c’è un notevole incremento della fibrosi midollare. C’è un'unica alterazione molecolare che accomuna tutte è tre queste malattie mieloproliferative, si tratta di un’alterazione di una parte del recettore per l’EPO che si chiama JAK2. Il recettore per l’EPO, quando è presente EPO subisce una sorta di dimerizzazione con attivazione degli elementi intracellulari, in particolare del JAK2 che subisce una dimerizzazione. Questa alterazione è presente: • Presente nel 100% dei casi nella policitemia vera • Nella trombocitemia e nella mielofibrosi è presente nel 50% dei casi In questi soggetti è presente una mutazione puntiforme del gene jak2 ovvero una fenilalanina al posto della valina e questo è responsabile di un’attivazione costitutiva del JAK2, e quindi è come se il recettore dell’EPO fosse continuamente stimolato dall’EPO, anche in assenza di eritropoietina il recettore funziona sempre. Quindi adesso si è scoperto per quale motivo i pazienti con policitemia vera hanno un aumento del numero dei GR! Proprio perché il recettore dell’EPO è sempre in funzione e dà all’eritrone un segnale di continua proliferazione. Non sappiamo bene perché il 50% dei pazienti con mielofibrosi e il 50% dei pazienti con trombocitemia ha questa alterazione, in questi pazienti infatti questa alterazione non si traduce in una proliferazione della linea eritroide bensì in una proliferazione della linea megacariocitica o nella mielofibrosi. Mielofibrosi La mielofibrosi è una malattia mieloproliferativa della cellula staminale che determina: • Eritropoiesi inefficace • Iperplasia megacariocitaria • Aumento del rapporto dei granulociti immaturi rispetto a tutti i granulociti E’ una malattia abbastanza rara con incidenza 1 su 100 mila abitanti, tipica dell’età anziana ma ci sono anche una certa quota di pazienti giovani, non c’è differenza tra i sessi. La mielofibrosi è caratterizzata da una ematopoiesi inefficace, quindi il midollo produce ma alla fine nel sangue periferico spesso troviamo una citopenia. Si ritrovano una fibrosi midollare (da cui il nome mielofibrosi) e una metaplasia mieloide, ovvero c’è una localizzazione extramidollare della ematopoiesi, soprattutto nella milza. Queste sono le malattie in cui troviamo le più grosse milze che potremmo mai vedere in qualunque patologia, nei pazienti con mielofibrosi la milza arriva in fossa iliaca, questo dovuto appunto alla metaplasia mieloide cioè la milza (ma anche il fegato) riacquista la funzione ematopoietica che aveva nella vita fetale → però si tratta di una ematopoiesi inefficace, cioè questa non è una produzione capace di sostituire il midollo e questa grossa milza alla fine crea ancora più danno perché: • Da un lato determina un sequestro splenico • Dall’altro lato l’aumento della milza determina la possibilità di cosiddetti infarti splenici, a causa della discrepanza tra l’irrorazione della milza e la sua grandezza. Ci sono diverse citochine implicate nella patogenesi, la più importante è la TGF-beta ma anche il platlet growth factor. Clinica La clinica è caratterizzata dal fatto che si tratta di una sindrome mieloproliferativa, quindi il paziente ha i segni di questo aumentato turn-over cellulare che si manifesta con: • Perdita di peso • Sudorazione notturna • Cachessia • spesso anemia • marcata splenomegalia, che comprime lo stomaco è dà una precoce sazietà, poiché riduce la capacità di espansione dello stomaco * Una delle caratteristiche cliniche più rilevanti – facendo una corretta anamnesi - nei pazienti splenomegalici è dovuta proprio al fatto che quando mangiamo hanno un senso di sazietà precoce perché la milza preme sullo stomaco e ne riduce la capacità di espansione. Diagnosi di laboratorio • Anemia Hb<10 g/dl presente nel 45% dei pazienti • Globuli bianchi <30000/ml usualmente bassi ma ci sono pazieti con globuli bianchi alti perché si tratta sempre di una sindrome mieloproliferativa per cui anche se la mielopoiesi nella maggior parte dei casi è inefficace a volte sono più presenti i segni di una mieloproliferazione piuttosto che di una citopenia • La piastrinopenia è presentente nel 26% dei casi • Vi possono essere alterazioni della coagulazione, caratterizzate dal fatto che questi pazienti sono più predisposti a sviluppare trombosi del circolo splacnico. Non si conosce bene il motivo per cui questi pazienti sviluppano trombosi solo nel circolo splacnico (vena porta, vena cava) tutto questo poi può comportare una compromissione epatica e di conseguenza delle alterazioni coagulative. Quindi noi spesso troviamo delle alterazioni coagulative che non sono tanto parte della malattia stessa quanto piuttosto una conseguenza della compromissione epatica legata a trombosi del circolo splacnico. Ci può essere la Sindrome di Budd-Chiari ovvero la sindrome da ostruzione delle vene sovra-epatiche, per cui il fegato viene a soffrire. Quadro periferico: Il quadro periferico è caratterizzato della presenza di un quadro leucoeritroblastico, cioè noi troviamo nel sangue periferico: • Sia elementi mieloidi immaturi cioè i meta mielociti o granulociti immaturi • Sia anche la presenza di elementi nucleati della serie rossa, gli eritroblasti • Accanto a questi ci sono le tipiche emazie a lacrima caratteristiche di questa patologia Se facciamo un aspirato midollare abbiamo la cosiddetta punctio-sicca ??, proprio perché la fibrosi midollare impedisce di aspirare il midollo, per cui la diagnosi deve essere fatta attraverso la biopsia osteo-midollare, una sorta di carotaggio dell’osso che riesce a prendere sia l’osso che il midollo e riesce ad evidenziare se ci sono queste turbe nel midollo associate ad una spiccata fibrosi midollare. Diagnosi Per il sospetto di diagnosi ci sono dei criteri identificati dall’OMS e sono: • Criteri clinici: epastosplenomegalia • Criteri ematologici: anemia- alterazione del numero dei globuli bianchi e delle piastrine o in difetto o in eccesso. • Caratteristiche morfologiche: la presenza di elementi immaturi sia della serie bianca che della serie rossa nel sangue periferico, una aniso-poichilocitosi e la presenza delle emazie a lacrima. • Nell’esame del midollo osseo avremo marcate fibrosi, cellularità che nella maggior parte dei casi è ridotta, ma può essere anche aumentata. Altre alterazioni morfologiche appannaggio soprattutto degli anatomo-patologi. Criteri maggiori Per porre la diagnosi di mielofibrosi occorre che ci siano queste proliferazioni megacariocitiche associate ad atipia, ma che ci sia una condizione in cui se noi troviamo la dimostrazione della mutazione di jak2 – in assenza di una diagnosi di policitemia vera – tutto questo configura un criterio maggiore per la diagnosi mielofibrosi Criteri minori: • Leucoeritroblastosi • Anemia • Palpabile epatomegalia • Aumento LDH, conseguenza del turn-over cellulare e della mielopoiesi inefficace In realtà per porre la diagnosi la WHO richiede che siano soddisfatti tutti e 3 i criteri maggiori (proliferazioni megacariocitiche associate ad atipia, assenza diagnosi di policitemia e dimostrazione mutazione di jak2) e 2 criteri minori. Ricordiamo che vi possono essere delle complicazioni, legate alle trombosi del circolo splacnico: • Ipertensione portale • Ascite • Varici esofagee • Rottura e infarti di milza Prognosi E’ una malattia che colpisce soprattutto i soggetti anziani ma anche a volte i giovani. La prognosi è stimata intorno ai 3-6 anni! Indicatori di prognosi avversa sono: • Età avanzata • Anemia • Presenza di stato iper catabolico • Trombocitopenia <100.000 • Leucopenia • Alterazioni citogenetiche Cause di morte: • Infezioni • Infarto cardiaco • Eventi trombo-emorrargici • talvolta anche una Trasformazione in leucemia acuta (un po’ come succede per la LMC) Terapia Purtroppo non abbiamo al momento terapie molto efficaci nei confronti di questo tipo di malattia, è possibile fare: 1. Trattamento palliativo 2. Riduce la proliferazione midollare con INF Ci sono numerosi farmaci in sperimentazione, soprattutto sono stati sviluppati anti-Jak2 che sembra stiano dando dei risultati interessanti Trombocitemia essenziale Un’altra malattia mieloproliferativa importante è la trombocitemia essenziale, dove appunto la proliferazione interviene soprattutto a carico dei megacariociti. Questa è una malattia leggermente più frequente con una incidenza di 2.5 casi per 100 mila abitanti, interessa soprattutto i pazienti anziani ma tra i soggetti < 40 anni interessa soprattutto le giovani donne. Diagnosi La diagnosi non è facile perché non vi sono dei criteri diagnostici ben definiti e molte volte si fa una diagnosi di esclusione. I criteri che dobbiamo considerare sono: 1. Piastrinosi > 600.000 2. Incremento dei megacariociti nel midollo osseo Come accennato sono più numerosi i criteri di esclusione: • Assenza di una diagnosi di policitemia vera • Assenza di una diagnosi di LM cronica • Assenza di una diagnosi di mielofibrosi • Assenza di una diagnosi di sindrome mielo-displastica • Assenza di altre cause che possono determinare una trombocitosi reattiva che sono le infiammazioni o le infezioni o le neoplasie. Ricordiamo che una condizione di piastrinosi si può avere: infiammazioni- neoplasie- splenectomia, perché se il paziente è stato splenectomizzato, in questo caso viene a mancare la sede di distruzione delle piastrine con relativo aumento. La WHO di recente, visto che nella trombocitemia è stata trovata l’alterazione del JAK2, questo ha permesso di abbassare ulteriormente il valore soglia per cui si considera la piastrinosi come sospetta per trombocitemia essenziale, come detto prima era un valore di 600 mila ora si è passati a un valore superiore a 450000. Quindi in presenza di > 400 mila piastrine, in presenza di alterazioni midollari dove c’è proliferazione megacariocitica e in assenza dei criteri di esclusione visti prima, e in presenza di questa anomalia molecolare che è il JAK2 → questo ci permette di fare diagnosi di trombocitemia essenziale anche con un valore di piastrine (400 mila) che prima era considerato non sufficiente per sospettare la trombocitemia essenziale. Caratteristiche cliniche: La maggior parte sono asintomatici, facciamo diagnosi perché abbiamo un reperto accidentale di piastrinosi. Alcuni pazienti si presentano con sintomi vasomotori, eventi trombo embolici, molto raramente eventi emorragici o ambedue, sia eventi trombotici che emorragici. * In realtà si pensa sempre che una piastrinosi non possa dare luogo ad un emorragia, tuttavia quando le piastrine sono molto elevate (almeno superiore a 1 milione e mezzo!), vi può essere un consumo del fattore di Von Willebrand, quindi è come se questi pazienti sviluppassero una malattia di Von Willebrand, cioè una malattia in cui vi è un difetto coagulativo con il risultato di una sindrome emorragica piuttosto che trombotica. Sintomi vasomotori → a causa di queste piccole trombosi il paziente accusa mal di testa, disturbi visivi (amaurosi fugace), disturbi auricolari, parestesie delle punte di mani e piedi ovvero un problema di “cattiva circolazione” del microcircolo legata a questo eccesso di piastrine. Policitemia vera La Policitemia Vera è una malattia mieloproliferativa in cui aumentano i globuli rossi e anche in questo caso, la diagnosi differenziale con la poliglobulia non è facile e non lo era prima della scoperta dell’anomalia molecolare del JAK2. Infatti se consideriamo la WHO del 2001, per fare diagnosi di policitemia vera i criteri fondamentali erano: • la valutazione della massa dei globuli rossi, valutazione che si faceva in medicina nucleare ed era una diagnosi molto complessa e anche poco attendibile. Ma la valutazione della massa dei GB era considerata uno dei criteri più importanti nella diagnostica della policitemia. • l’assenza di cause secondarie di poliglobulia, assenza delle condizioni familiari, assenza delle condizioni che determinano un aumento di eritropoietina endogena (ipossia), produzione inappropriata di eritropoietina da parte di alcuni tumori (in particolare il tumore renale). • un’altra caratteristica clinica era considerata la splenomegalie, poiché quasi tutte le malattie mieloproliferative si caratterizzano per la splenomegalia • assenza di alterazioni cromosomiche • un altro criterio importante è definito dalla formazione di colonie eritroidi endogene, cioè non stimolate dall’eritropoietina, cosa vuol dire? In condizioni normali per ottenere delle colonie eritroidi dal midollo o dal sangue di un paziente normale, dobbiamo metterci l’EPO, cioè prendiamo un midollo normale con delle cellule staminali, lo mettiamo in coltura e mettiamo l’EPO così da quel midollo possiamo sviluppare le colonie eritroidi. Nei pazienti con policitemia vera le colonie eritroidi si possono sviluppare anche senza mettere l’EPO, si parla di formazione endogena di colonie eritroidi. Criteri minori sono: • Aumento delle piastrine > 400 x 10^9 • Leucocitosi 12 x 10^9 • Biopsia del MO con la prominente iperplasia eritroide • ! Bassi livelli di EPO → questo ci permette facilmente di fare diagnosi differenziale tra le condizioni di poliglobulia date da un aumento di eritropoietina, rispetto alla policitemia vera. Perché tutte le condizioni che abbiamo visto (ipossia da elevate altitudini, fumatori) provocano un poliglobulia secondaria ad ipersecrezione di EPO. Nella policitemia vera dove c’è invece un recettore per l’EPO che è costitutivamente attivato e vi è l’aumento del numero dei GR, chiaramente il meccanismo di feedback fa sì che non viene prodotta eritropoietina poiché c’è già una quantità notevole di globuli rossi, l’organismo non produce EPO. Il dosaggio sierico dell’eritropoietina è uno degli spartiacque che noi utilizziamo quando abbiamo un paziente con poliglobulia e non sappiamo se è secondaria o primitiva. Però visto che adesso abbiamo questa informazione, cioè che tutti i pazienti o comunque quasi tutti i pazienti affetti da policitemia vera presentano la mutazione JAK2, questo è diventato il criterio maggiore per fare la diagnosi!! Non c’è più bisogno di andare a fare in medicina nucleare la valutazione della massa eritroide, adesso basta trovare: • Hb > 18,5 nell’uomo e > 16,5 nella donna • Mutazione JAK2 La diagnosi si può fare in presenza di questi due criteri maggiori e di almeno 1 di quelli minori oppure 1 criterio maggiore e 2minori. • Ipercellularità del midollo con prominente iperplasia di tipo eritroide • Valori di EPO inferiori alla norma • Formazione di colonne eritroidi senza la somministrazione di EPO Quindi: • 2 criteri maggiori e 1 minore • 1 criterio maggiore e 2 minori A volte si usa 1 solo criterio maggiore e uno minore, perché il JAK2 in effetti è mutato in tutti i pazienti con policitemia vera, però non sempre la mutazione avviene nel 617V, ma a volte la mutazione avviene in un altro punto del gene → per cui facendo la valutazione molecolare della mutazione in questo punto del gene (617V) a volte non si riesce a trovarla perché l’aminoacido mutato è un altro. Il concetto comunque è sempre lo stesso, qualunque mutazione del gene JAK2 determina un’attivazione costitutiva del recettore per l’EPO, così l’eritrone è sempre stimolato a crescere e a produrre GR. Quindi in realtà questo tipo di mutazione la ritroviamo nel 90% dei pazienti con policitemia vera e non nel 100%. Perché muoiono i pazienti con policitemia vera? Perché come in tutte le malattie mieloproliferative, in particolare la mielofibrosi ma anche la trombocitemia (seppure in minor misura) e in una situazione intermedia la policitemia vera, questi pazienti possono andare incontro a: • Trombosi, soprattutto del circolo splacnico, cerebrali 30% • Leucemia/linfoma 15% • Patologia non maligna • Emorragie • Trasformazione di questa malattia o in mielofibrosi (fibrosi post-policitemia) oppure addirittura in leucemie acute, sono condizioni rare Alcune volte si può passare ad una “fase spenta” di malattia, il paziente passa ad una condizione di poliglobulia e man mano diventa anemico. Ciò è dovuto al fatto che man mano si accumulano alterazioni molecolari a carico delle cellule staminali che trasformano questa malattia, da una malattia mieloproliferativa normale a una condizione di mielodisplasia e addirittura una mielodisplasia ipocellulare. Liberazione di citochine che determinano fibrosi midollare. Quindi questi pazienti passano da una condizione in cui per terapia bisogna togliere l’eccesso di GR, ad una condizione in cui vi è richiesta di GR. Terapia 1. Farmaci* (idrossiurea) che inibiscono la proliferazione midollare 2. Salasso, utilizzato più frequentemente, anche se preferiamo fare l’eritroaferesi. L’eritroaferesi è una procedura per cui invece di togliere tutto il sangue togliamo solo i GR, così non depauperiamo il paziente degli altri costituenti utili del plasma ma anche ci permette di togliere un maggior numero di GR. Fondamentalmente il concetto è sempre lo stesso: togliere l’eccesso di GR e mantenere l’ematocrito a valori che non siano superiori a 45-48%. La letteratura non è concorde o meglio tutti gli Autori sono d’accordo che bisogna mantenere l’ematocrito sotto il 48% , qualcuno dice anche sotto il 45%, però fondamentalmente l’obiettivo è quello, da raggiungere come visto o con i farmaci o con i salassi e l’eritroaferesi. * Si è visto che probabilmente se si utilizzano dei farmaci come l’idrossiurea o altri farmaci, questo può in qualche modo favorire il rischio di trasformazione della policitemia vera in una malattia neoplastica tipo leucemia mieloide acuta, allora si preferisce fino a quando è possibile tenere i pazienti con una terapia a base di salassi, soprattutto se si tratta di pazienti giovani, chiaramente se si tratta di pazienti anziani non ci poniamo questo problema perché l’effetto mutageno di questi farmaci, ammesso che vi sia, è un effetto che interviene dopo decine di anni. Benefici: • Maggiore sopravvivenza • Riduzione incidenza di cancro/leucemia/linfoma Rischi: • Il rischio maggiore è la trombosi (primi 3anni), quindi è fondamentale somministrare l’aspirina per ridurre il rischio di eventi trombotici. • Accelerazione della progressione in mielofibrosi • Non controllata splenomegalia-iperuricemia-prurito Quindi il nostro obiettivo principale in questi pazienti con policitemia vera e in generale in tutti i pazienti con malattie mieloproliferative è quello di ridurre il rischio di questi pazienti di sviluppare la trombosi e in questo senso l’aspirina è uno dei capisaldi della terapia, poiché essendo un antiaggregante piastrinico non permette la formazione di trombi. Ci sono ricordiamo delle sperimentazioni con farmaci anti-JAK2. Prognosi La policitemia vera è una malattia abbastanza benigna seppure si tratta di una proliferazione clonale della cellula staminale, quindi sul piano biologico di una neoplasia, sul piano clinico basta mantenere l’ematocrito al di sotto di determinati valori ( 45-48% ) che si riduce il rischio della malattia di evolversi. Chemioterapia benefici: • Ridotta incidenza di trombosi (profilassi con l’aspirina) • Generalmente tollerata • Controllo dei sintomi legati all’eccessiva proliferazione (pazienti con sudorazione nottura, perdita di peso, febbricola – questi vanno trattati con la chemioterapia non con il salasso) Si utilizza l’IDROSSIUREA un chemioterapico ben tollerato, tra tutti è quello con il minore potere mutageno, tanto che viene utilizzato anche nei bambini affetti da anemia falciforme, patologia in cui c’è un eccesso di HbS. L’HbS tende a precipitare in condizioni di ipossia e acidosi e provoca deformità dei GR, fatti microtrombotici e tutta la sintomatologia che abbiamo trattato. Si è visto che l’idrossiurea aumenta la produzione di HbF ovvero fetale, quindi se in questi bambini aumenta l’HbF, si riduce percentualmente l’HbS e quindi c’è un significativo miglioramento dei sintomi. Finora gli studi fatti e i follow up a 15anni hanno dimostrato che in questi bambini non si sviluppano leucemie secondarie, a dimostrazione che l’idrossiurea è un farmaco con basso potere mutageno. E’ chiaro che bisogna ancora aspettare del tempo, ci vogliono più di 20 anni perché molto spesso questi farmaci diano luogo a mutazioni tali da determinare una trasformazione leucemica anche se gli studi sono confortanti. Rischio: Non tutti utilizzano l’idrossiurea ma farmaci con un alto potere mutageno, certamente questi agenti possono incrementare il rischio di leucemia. Ricapitolando: 1. Policitemia vera c’è un aumento del nuemro dei globuli rossi 2. Trombocitemia essenziale in cui c’è un aumento del numero delle piastrine 3. Mielofibrosi idiopatica Tutti e 3 sono delle patologie mielo-proliferative croniche che hanno in comune la mutazione a livello del JAK2 e un elevato rischio trombotico. Lezione 15 Il Processo Emostatico Il processo di emostasi si può dividere in tre fasi. La fase iniziale in seguito al danno endoteliale è quella del vaso spasmo,successivamente abbiamo la fase del coagulo piastrinico e infine la fase di formazione del coagulo di fibrina. E' chiaro che a questo punto ci sono delle alterazioni specifiche delle tre diverse fasi. Ci può essere un'alterazione della fase iniziale,della cosiddetta fase vasculo-piastrinica,e questo può essere dovuto al fatto che il tessuto di sostegno dei vasi è alterato. Questo succede in alcune patologie congenite del collagene,oppure in seguito ad alterazioni vasali da vasculite. Oltre alle alterazioni dei vasi e del tessuto perivascolare ci può essere un difetto piastrinico. Queste nel loro insieme costituiscono le alterazioni della fase vasculo-piastrinica. Da un punto di vista clinico avremo: 1. soprattutto le ecchimosi e le petecchie 2. stillicidi dalle mucose 3. prolungato sanguinamento dopo un intervento chirurgico. In seguito abbiamo la fase della coagulazione vera e propria e quindi tutte quelle patologie in cui c'è un difetto dei fattori coagulativi. In questo caso avremo come sintomatologia clinica sopratutto gli emarti,gli ematomi ed emorragie interne. Questi sintomi dipendono appunto dai fattori della coagulazione. Ci possono essere dei difetti nella stabilizzazione del coagulo. Come sapete nella fase finale interviene il fattore XIII che serve alla stabilizzazione del coagulo,se c'è un difetto a questo livello al momento della nascita si osserverà una prolungata emorragia al moncone del funicolo ombelicale e un ritardo nella cicatrizzazione delle ferite. Ci possono essere difetti nella lisi del coagulo e in questo caso la presentazione clinica consisterà in prolungate emorragie dalle ferite chirurgiche o da trauma. Vediamo più nel dettaglio questi meccanismi. Nella prima fase abbiamo uno spasmo muscolare,questo meccanismo interviene nell'arco di pochi millisecondi. E' legato all'attivazione dei recettori del dolore ed alla lesione del tessuto muscolare liscio. Questo fondamentalmente serve per dare il tempo alle piastrine ed ai fattori della coagulazione di intervenire. Le piastrine sono dei piccoli frammenti dei megacariociti. La normale conta è tra le 130.000 e 400.000 piastrine. Hanno il compito di: secernere alcuni fattori della coagulazione,secernere fattori che possano riparare il danno endoteliale e costituire il trombo piastrinico che è una sorta di “tappo iniziale” formato da sole piastrine. Inoltre sono in grado di richiamare in quella sede i globuli bianchi,che creano un ambiente infiammatorio locale che favorisce la formazione dell'ulteriore coagulo. Vista in sezione longitudinale,la piastrina appare come una micro particella molto piccola e molto complessa,che contiene al suo interno una serie di organuli necessari per la propria funzione. Dicevamo che le piastrine sono derivanti dai megacariociti,che sono delle cellule particolarmente grandi,con una spiccata poliploidia e che determinano la produzione di un numero elevato di piastrine. Una delle caratteristiche delle piastrine è la loro capacità di aderire al fattore di von Willerbrand . Il fattore di von Willerbrand serve a legare le piastrine tra di loro,ma soprattutto a legarle al subendotelio o all'endotelio,per cui è importante nelle prime fasi della coagulazione proprio per fare aderire queste piastrine alla lesione endoteliale. Le piastrine,una volta a contatto con il sub endotelio,vanno incontro a cambiamenti della loro morfologia che permettono l'aggregazione piastrinica e la loro coalescenza. Vi dicevo che le piastrine sono strutture complesse che comprendono molti granuli,a loro volta suddivisi in granuli densi,alfa granuli e lisosomi. Dentro questi granuli ci sono sostanze che servono: 1 alla funzione piastrinica,per cui alcune di queste sostanze serviranno all'aggregazione piastrinica, 2 alla coagulazione del sangue Contengono inoltre enzimi idrolitici e tutto un laboratorio molto importante per la funzione piastrinica. Lasciamo perdere i vari nomi e le varie funzioni,quello che è importante è che durante l'attivazione, da questi granuli si liberano delle sostanze ad effetto procoagulante. La più importante di queste sostanze è il trombossano A2 (e anche il trombossano B2).Il trombossanoA2 è il più importante,deriva dal metabolismo dell'acido arachidonico. I vari farmaci che interferiscono con la funzione piastrinica vanno a bloccare proprio la produzione del trombossanoA2.Il più importante tra questi farmaci è l'aspirina,che è il più importante anti-aggregante piastrinico. Accanto all'aspirina ci sono l'indometacina e il fenilbutazone che sono tutti farmaci anti infiammatori non steroiodei (FANS), che hanno la stessa funzione anti aggregante proprio perchè interferiscono con la produzione di trombossano. La differenza principale tra l'asprina e gli altri farmaci è che l'aspirina,una volta che si lega alla piastrina,interferisce col suo metabolismo in maniera permanente,mentre gli altri FANS inibiscono la funzione piastrinica soltanto per il tempo in cui si trovano in circolo nel plasma. Nel caso degli altri FANS la durata dell'inibizione è proporzionale alla loro emivita plasmatica mentre con l'uso dell'aspirina ogni piastrina, che ha un emivita di 5-7gg, non sarà più attiva. Quindi è per questo che ci vogliono 5-7gg per annullare l'effetto anti aggregante dell'aspirina. Questo significa che se un paziente deve essere sottoposto a intervento chirurgico,in cui è necessaria una buona funzione aggregante piastrinica,occorre sospendere l'aspirina dai 5 ai 7 gg prima all'intervento chirurgico. Per gli altri farmaci tutto dipende dalla loro emivita,che usualmente è di 12-24 h e dunque basta sospendere il farmaco 24 ore prima dell'intervento. Poi ci sono farmaci che invece stimolano l'attività piastrinica e che noi usiamo per andare a vedere la funzionalità delle piastrine,per fare il test dell'aggregazione piastrinica. Un plasma ricco di piastrine viene messo in un sistema che ne rivela gli aggregati,per cui una volta che il plasma viene esposto a sostanze che stimolano l'aggregazione piastrinica,come l'ADP o l'epinefrina,questo determina un'alterazione dell'onda primaria e secondaria rilevata da questo sistema e ci permette di valutare la velocità e l'entità dell'aggregazione piastrinica. Questo permette di valutare non solo semplicemente il numero ma anche la funzionalità delle piastrine. Le piastrine possono avere una ridotta funzione perchè: 1. sono poche 2. in numero normale ma non funzionali. Quindi le anomalie a carico delle piastrine possono essere numeriche o funzionali. Le anomalie numeriche possono essere nel senso di un aumento di numero o di una diminuzione. Già ieri abbiamo visto che le cause di aumento della conta piastrinica sono le malattie mieloproliferative, quali la trombocitemia essenziale ma anche altre come la LMC, la mielofibrosi idiopatica, la policitemia vera. Queste patologie possono determinare un aumento della conta piastrinica. Esistono delle condizioni in cui le piastrine aumentano in maniera secondaria,non per malattia linfoproliferativa. Queste condizioni sono date dall'infiammazione,dall'emorragia,dall'anemia emolitica(quando parlo di emorragie mi riferisco anche alle anemie sideropeniche secondarie ad emorragie). E' molto frequente trovare pazienti con un quadro di piastrinosi che in realtà hanno un'anemia sideropenica. Anche le neoplasie,la splenectomia,lo sforzo fisico (in maniera molto relativa) possono determinare piastrinosi. Le condizioni di piastrinopenia possono essere dovute: 1. a mancata produzione, 2. a diluizione, 3. a un'alterata distribuzione, 4. ad aumentato consumo. Le cause più importanti sono quelle da alterata produzione e da aumentato utilizzo; le cause diluizionali e da alterata distribuzione sono più rare,meno importanti sul piano clinico. Per trombocitopenia definiamo una quantità sub normale di piastrine nel sangue. Dicevo che le cause possono essere:eccesso di distruzione,alterata distribuzione,diminuita produzione,aumentata diluzione e poi abbiamo una condizione che può essere definita come pseudo-trombocitopenia. Questa è una condizione non particolarmente rara. I pazienti giungono all'osservazione perchè dopo aver fatto un emocromo hanno un riscontro occasionale di un numero molto basso di piastrine. In realtà questi pazienti non hanno alcuna manifestazione emorragica e non hanno neanche storia di emorragie. Quando noi facciamo un emocromo prendiamo il sangue del paziente e lo mettiamo in una provetta con dell'anticoagulante. Normalmente questo anti-coagulante è l'EDTA. Può succedere che alcuni pazienti o per la presenza di alcuni autoanticorpi,o per un difetto delle piastrine,non sono sensibili all'effetto anticoagulante dell'EDTA ,per cui queste piastrine in provetta si aggregano e la macchina ne conta di meno. Basta rifare l'esame con eparina o con citrato oppure procedere immediatamente alla conta piastrinica prima che il sangue abbia il tempo di coagulare. La condizione di alterata distribuzione è meno importante come frequenza e dal punto di vista clinico,è dovuta sostanzialmente ad ipersplenismo. Vi dicevo che nella mielofibrosi e in alcune epatopatie c'è una milza aumentata di volume e quindi si verifica un sequestro splenico,senza che vi sia una riduzione importante della conta piastrinica. Ci sono però delle condizioni in cui c'è un deficit di produzione piastrinica,allora la riduzione della conta sarà molto più importante. Sono delle forme in cui è ridotta la funzione midollare,come l'anemia di Fanconi,che è una condizione congenita di alterazione dell'ematopoiesi; l'emoglobinuria parossistica notturna; in tutte quelle condizioni che si associano ad aplasia midollare,infezioni virali,deficit di vitamina B12 e di folati; alcuni farmaci come i chemioterapici,oppure in caso di invasione midollare da parte di un processo neoplastico (leucemie acute,quando l'invasione del midollo da parte dei blasti impedisce la proliferazione dei megacariociti con ridotta immissione in circolo di piastrine). Ma la condizione più frequente che causa piastrinopenia è la distruzione periferica delle piastrine. Fondamentalmente c'è un consumo eccessivo di piastrine che non riesce ad essere compensato dalla produzione midollare. Le condizioni in cui questo avviene sono :la porpora trombocitopenica idiopatica o morbo di Werlhof indicato dalle nuove linee guida come porpora immune. Nei pazienti affetti da HIV esiste una forma particolare di Porpora Trombocitopenica Idiopatica che è probabilmente legata al fatto che c'è una sorta di cross-reattività degli anticorpi diretti contro le piastrine col virus dell'HIV. Ci possono alcuni farmaci che provocano piastrinopenia. Il primo fra tutti è l'eparina. E' una condizione strana e grave nello stesso tempo. L'eparina, voi sapete, viene utilizzata come anticoagulante nei pazienti che hanno avuto ad esempio una trombosi. Un paziente in terapia con eparina e contemporanea riduzione del numero delle piastrine potrebbe però sviluppare importanti emorragie. E' una condizione difficile da trattare. Un'altra condizione difficile da trattare è la cosiddetta coagulazione intravascolare disseminata (CID). Allo stesso modo complicato è il trattamento della porpora trombotica trombocitopenica o sindrome di Moschowitz. TROMBOCITOPENIA ASSOCIATA AL TRATTAMENTO CON EPARINA Condizione grave ma per fortuna rara. Soprattutto adesso con le nuove eparine a basso peso molecolare è una condizione che si riscontra molto meno frequentemente. La patogenesi è legata alla produzione di anticorpi contro l'eparina che cross-reagiscono con le piastrine e l'endotelio. L'interessamento endoteliale complica ulteriormente la diatesi trombotica perché la lesione endoteliale aumenta il rischio trombotico. Il paziente è contemporaneamente a rischio di sviluppare trombosi o emorragie. Questi pazienti possono manifestare: trombosi venose profonde, embolie polmonari, gangrene, trombosi cerebrali ed arteriose, infarti delle ghiandole surrenali, lesioni cutanee (dovute a trombosi dei vasi superficiali del derma). PORPORA TROMBOCITOPENICA IDIOPATICA O MORBO DI WERLHOF E' sempre legata alla presenza di anticorpi che si legano alle piastrine. I linfociti B attivati in maniera anomala producono anticorpi contro le piastrine. Il complesso piastrina-anticorpo viene riconosciuto dai macrofagi e dal sistema reticolo-endoteliale ed eliminato. E' poco frequente ed interessa in particolare le donne in giovane età. La sintomatologia è spesso preceduta una infezione virale. Dunque l'ipotesi patogenetica presuppone la produzione di anticorpi contro il virus che per mimesi molecolare (somiglianza tra antigeni virali ed antigeni presenti sulle piastrine. La diagnosi è in genere di esclusione. La presenza di anticorpi anti-piastrine è assolutamente aspecifica. E' stato valutato che solamente il 20-30% dei pazienti ha la presenza di anticorpi anti-piastrine e quindi non abbiamo test specifici e sensibili per accertare la diagnosi. Questi pazienti hanno una piastrinopenia isolata con MPV aumentato perché le piastrine più giovani sono più grandi (la distruzione piastrinica determina un aumento nel turnover midollare). I moderni contatori cellulari calcolano il numero di piastrine reticolate che sono equivalenti ai reticolociti della serie eritroide. Le prove coagulative sono normali in questi pazienti. Prima si riteneva che nel midollo il numero di megacariociti dovesse essere aumentato in compenso all'aumento nella distruzione piastrinica. In realtà si è poi visto che il numero di megacariociti midollari è quasi sempre normale perché gli anticorpi si legano ai megacariociti midollari e ne determinano la distruzione. Non tutti i pazienti devono essere trattati. Non vanno trattati i pazienti con più di 50000 piastrine (noi in realtà iniziamo il trattamento a 30000) e senza manifestazioni emorragiche. Vanno trattati i pazienti con un numero di piastrine inferiore a 30000 e/o con manifestazione emorragiche. Il trattamento deve mirare a ridurre il numero di anticorpi e dunque sarà un trattamento immunosoppressivo. Il trattamento di prima scelta sarà dunque il prednisone che è uno steroide. Se il paziente non risponde al trattamento si può ricorrere all'iniezione endovena di immunoglobuline. Queste immunoglobuline vanno a legarsi al frammento Fc del macrofago ed in pratica vanno ad impedire la fagocitosi del complesso piastrina-anticorpo. Con analogo meccanismo agiscono le Ig anti-D(non sono sicuro di questo purtroppo non si capisce). Nei casi refrattari alla terapia medica si ricorre alla splenectomia (sede di maggiore distruzione). In realtà oggi possiamo utilizzare altri due presidi terapeutici. Uno è rappresentato dal Rituximab (anticorpo monoclonale anti-CD20). L'altro dall'utilizzo di peptidi antagonisti dei recettori per la trombopoietina; ne esistono due di cui uno sta per entrare in commercio. Sono farmaci che mimano la presenza di trombopoietina e dunque stimolano la produzione di piastrine. In condizioni di urgenza si ricorre alla splenectomia d'urgenza o alla trasfusione diretta di piastrine(non indicata nella normale terapia). C'è una peculiarità in questa patologia. I bambini che hanno questa malattia quasi sempre vanno in remissione,spesso spontanea (senza cortisone) e duratura. Gli adulti di solito rispondono alla terapia ma la remissione è quasi sempre temporanea (da due mesi a qualche anno)e spesso si hanno recidive (ITP recidivante o cronica). Oltre alle piastrinopenie si possono verificare delle piastrinopatie (piastrine che funzionano in maniera anomala). Le piastrinopatie possono essere acquisite (chi fa terapia con aspirina ha una piastrinopatia) o congenite. L'insufficienza renale cronica con uremia determina piastrinopatia. Le paraproteinemie (mieloma) possono causare piastrinopatia per interferenza di queste proteine con i normali antigeni piastrinici. Alcune malattie linfoproliferative possono dare sintomi da deficiente funzionalità piastrinica perché tali piastrine originano da un clone neoplastico ed avranno delle anomalie nel funzionamento. Glicogenosi ed interventi in circolazione extracorporea sono situazioni in grado di interferire con il funzionamento piastrinico. Le forme congenite possono essere legate a difetto: • dell'adesione piastrinica per deficit del recettore per il fattore Von Willebrand • dell'aggregazione piastrinica :tromboastenia di Glanzmann in cui manca il recettore per il fibrinogeno e sindrome di Bernard- Soulier in cui manca il recettore GpIb. Inoltre ricordate che una volta attivate, le piastrine rilasciano tutta una serie di sostanze contenute nei granuli citoplasmatici. Se tali sostanze sono deficitarie si avranno dei problemi nella progressione del processo emostatico. Esistono infatti difetti riguardanti: • lo storage piastrinico o storage pool disease • la secrezione piastrinica o aspirin like disease • l'attività procoagulante piastrinica (nel processo della coagulazione l'attività piastrinica è importante nella produzione di alcuni fattori della coagulazione e nella catalizzazione del processo stesso). La coagulazione è il processo principale nel contenere le perdite sanguigne. Deve essere veloce e precisa. Fondamentalmente consiste nella conversione del fibrinogeno in fibrina. I fattori della coagulazione sono presenti nel plasma e sono prodotti in maniera inattiva dal fegato (il fattore VIII prodotto anche dall'endotelio. Un fattore attiva l'altro con meccanismo a cascata, la famosa cascata della coagulazione. La cascata della coagulazione può essere attivata per via intrinseca o per via estrinseca. La via estrinseca della coagulazione viene attivata, ad opera di un fattore tessutale, TF (la cosiddetta tromboplastina tessutale o fattore III) presente nei tessuti e da questi esposto in seguito ad un danno cellulare. Viene detta estrinseca proprio perché il fattore tissutale è estrinseco al sangue. La via intrinseca prende origine da proteine presenti nel sangue che vengono attivate in seguito a stimoli di varia natura, tra cui la degranulazione piastrinica. Per fare funzionare sia la via intrinseca che quella estrinseca un fattore molto importante è il calcio. NB non dovete sapere tutta la cascata coagulativa ma dovete avere chiari alcuni concetti. Possiamo distinguere una fase iniziale, una intermedia e la fase finale in cui si forma il coagulo. FASE INIZIALE Come già detto la via estrinseca nella fase iniziale viene attivata dal fattore VII mentre la via intrinseca dai “fattori di contatto”. Il TF(fattore tissutale) lega specificamente una proteina plasmatica, il fattore VII e gli ioni calcio, formando un complesso dotato di attività enzimatica. Il complesso che risulta (COMPLESSO TFFVII-Ca ) è enzimaticamente attivo, possiede un’alta affinità per il fattore X e ne catalizza l’attivazione a Fxa (attivato). Il fattore Xa è in grado di attivare il fattore VII, che a sua volta attiverà altro fattore X. Si verifica una autoamplificazione che stimola fortemente il fattore X. Il fattore VII può attivare anche il fattore IX. 2+ La prima tappa della via estrinseca è rappresentata dalla attivazione del cosiddetto “sistema plasmatico attivabile da contatto”, costituito da 4 proteine: 1FATTORE DI HAGEMAN (HF o FATTORE XII) 2PRE-CALLICREINA PLASMATICA 3- CHININOGENO AD ALTO PESO MOLECOLARE (HMWK) 4- FATTORE XI o PTA (antecedente plasmatico della tromboplastina). Se noi abbiamo un deficit del fattore settimo abbiamo un'emorragia severa. Se invece abbiamo un deficit di fattore XII o XI non abbiamo emorragie. Il fattore di Hageman si chiama così perché fu scoperto in un paziente che si chiamava Hageman e che morì per embolia polmonare(fatto trombotico e non emorragico). I fattori iniziali della via estrinseca e della via intrinseca convergono sul fattore IX e X che rappresentano i fattori della fase intermedia. Il ruolo del FIXa consiste nella conversione del fattore X (o di Stuart) nella sua forma enzimaticamente attiva, Fxa. Il fattore Xa, interagendo con il fattore Va, i fosfolipidi (della membrana piastrinica, endoteliale, leucocitaria) e gli ioni calcio, forma un complesso multimolecolare che prende il nome di PROTROMBINASI o COMPLESSO PROTROMBINASICO, in grado di agire proteoliticamente sulla protrombina (fattore II) trasformandola in trombina. Tutto ciò converge sulla conversione del fibrinogeno in fibrina da parte della trombina. La formazione della fibrina non è altro che la stabilizzazione delle catene di fibrinogeno. Per formare il coagulo di fibrina il fibrinogeno subisce un clivaggio da parte della trombina. Tale clivaggio libera i fibrinopeptidi A e B (se ricercati nel plasma ci indicano che il fibrinogeno si è trasformato in fibrina). Livelli elevati di fibrinopeptidi A e B sono marcatori di fibrinogenesi. Il TF può anche attivare il fattore IX e ci sono molti punti di contatto tra via estrinseca e via intrinseca. Il fattore VII ed il IX attivano il fattore X con la complicità del fattore VIII. Il fattore X, con la complicità del fattore V, trasforma la protrombina in trombina che a sua volta trasformerà il fibrinogeno in fibrina. Il fattore X è il punto di contatto tra le due vie. Le piastrine hanno un ruolo importante in quanto fungono da catalizzatrici del processo della coagulazione e perché sulla loro membrana si realizzano importanti reazioni atte a favorire la coagulazione. Le piastrine legano i fattori V, VIII ed XI e li attivano. I fattori X, IX e II rappresentano i fattori della fase intermedia della coagulazione. La fase successiva all'attivazione della protrombina è quella dell'amplificazione. Succede un macello, si attiva il fattore VIII, V e XI e molti altri. La fase di propagazione è quella in cui piccole quantità di trombina adesso diventa una grande quantità di trombina. Ricapitolando in breve:l'emostasi ha inizio con l'interazione del TF con il fattore VII sulla superficie delle cellule endoteliali. La piccola quantità di trombina generata durante le fasi iniziali attiva localmente le piastrine, sulla cui superficie hanno luogo le reazioni successive. Il burst di trombina susseguente porterà alla formazione di un coagulo stabile di fibrina. Possiamo studiare il processo coagulativo mediante il PT (attività protrombinica) ed il PTT (tempo di tromboplastina parziale). L'attività protrombinica va a studiare la via estrinseca. Il PTT studia la via intrinseca. Dunque PT→via estrinseca e PTT→via intrinseca. Per la valutazione del PTT si aggiunge al plasma del paziente il caolino(sostituto della membrana fosfolipidica piastrinica favorendo la coagulazione). Il PTT studia i fattori della intrinseca XII, XI e VIII. La via comune (X, II, I) è studiata da entrambi i test. Viceversa l'attività protrombinica studia soltanto il fattore VII. In questo caso si aggiunge al plasma il TF e si valuta il tempo di coagulazione. Se abbiamo un paziente con allungamento dell'attività protrombinica dovremo sospettare un difetto del fattore VII. Viceversa in un paziente con allungamento isolato del PTT dovremo sospettare un deficit dei fattori XII, XI e VIII. Tuttavia non è così semplice. Il bleeding time si esegue facendo un taglio al paziente, sul braccio o sul padiglione auricolare, e valutando quanto tempo ci vuole affinché si formi il coagulo. Se un paziente ha un'alterazione della fase vasculo-piastrinica avrà il bleeding time allungato. In un paziente con allungamento isolato del PTT (piastrine normali, bleeding time normale e PT normale) cosa dobbiamo fare? Quello che si fa è prendere il sangue del paziente e mescolarlo, in rapporto 1:1, con il plasma normale. In questo caso ci può essere la correzione del PTT oppure no. Se si verifica la correzione bisogna distinguere ulteriormente se il paziente sanguina o non sanguina. Perché se non sanguina il paziente avrà un deficit dei fattori di contatto (abbiamo detto che in caso di deficit dei fattori di contatto non abbiamo emorragie). Se invece il paziente sanguina il difetto sarà nei fattori più bassi ovvero: fattore VIII, IX o XI (condizione di emofilia). Se il PTT non viene corretto dall'aggiunta di plasma normale dobbiamo sempre distinguere se il paziente sanguina o no. Se sanguina è perché il plasma normale non riesce a correggere il deficit di fattore VIII o IX perché nel plasma del paziente sono presenti anticorpi, che neutralizzano i fattori da noi aggiunti (emofilia acquisita). Se il paziente non sanguina invece siamo di fronte ad un falso positivo. Questo succede nel lupus anti-coagulant, nella sindrome da anticorpi anti-fosfolipidi ed anti-cardiolipina che provocano allungamento del PTT. Questo succede perché noi inizialmente aggiungiamo caolino( fosfolipidi) per fare avvenire la reazione che viene inattivato dalla presenza di questi anticorpi. Quindi l'allungamento del PTT, in questo caso, non dipende da deficit di alcun fattore ma dal fatto che la presenza di questi anticorpi blocca l'avvio della reazione. Questi pazienti avranno problemi di diatesi trombotica e questo può sembrare paradossale se si ignora tale possibilità diagnostica. Il processo coagulativo deve anche essere regolato negativamente altrimenti al primo taglio moriremmo di trombosi. Intanto il fegato distrugge i fattori di coagulazione e poi in ambito fisiologico c'è una bilancia tra fattori procoagulanti ed anticoagulanti. I fattori anticoagulanti sono: • le antitrombine, la più importante è l'antitrombina III • fattori inibitori del TF • sistema proteina C ed S. L'antitrombina III è molto importante in tale bilancia perché inattiva la trombina e tutte le proteasi (XII,XI, IX e X). Tutto tale sistema è accelerato dall'eparina. L'eparina agisce da anticoagulante perché accelera e potenzia l'attività dell'anti-trombina terza. E' importante il dosaggio dell'ATIII in un paziente che fa terapia con eparina. Se un paziente non ATIII potremo fargli chili di eparina senza avere nessun risultato. Quando avete un paziente che non risponde alla terapia con eparina la prima cosa che dovete fare è dosare i livelli dell'ATIII. Le proteine C e S inattivano i fattori V ed VIII. Una volta formato il coagulo e riparata la ferita questo deve andare in retrazione. La retrazione del coagulo è affidato al sistema della fibrinolisi. Il sistema della fibrinolisi è parallelo a quello della coagulazione solo che adesso il target finale è la trasformazione del plasminogeno in plasmina. La plasmina distrugge la fibrina generando i frammenti di degradazione della fibrina ed il famoso D-Dimero. Il dosaggio del D-Dimero è indice di fibrinolisi. Ci sono inibitori anche della plasmina, tra cui: PAI1 e l'alfa due antiplasmina. Vi ricordo che la determinazione del fibrinopeptide A è indice invece di fibrinogenesi. EMOFILIA E' una delle patologie coagulative più importanti. Parleremo di emofilia A legata a difetto del fattore VIII. E' una malattia legata al sesso, a trasmissione diaginica, in quanto il gene per il fattore VIII è sul cromosoma X. Saranno dunque in maggioranza colpiti i maschi in quanto a tutti gli effetti sono omozigoti per il cromosoma X (le donne hanno due cromosomi X e per essere malate dovrebbero ereditare entrambi i cromosomi X con l'alterazione genetica). Esistono diversi gradi di emofilia che dipendono dalla quantità di fattore VIII presenti nel plasma. Fino al 25% di fattore VIII rispetto al valore normale non si ha alcuna sintomatologia con sanguinamenti solo post-traumatici. Dal 25% al 6% è una condizione lieve. Dal 5% all'1% è una condizione moderata. Sotto l'1% si parla di emofilia severa. La mancanza del fattore VIII determina una drastica riduzione del fattore X perché il fattore VIII è necessario per attivare il fattore X. Sono colpite soprattutto le articolazioni con emorragie intracapsulari. Il paziente riferisce dolore e tumefazione all'articolazione che può successivamente infiammarsi con anchilosi articolari. Si possono avere ematomi muscolari. Tipico è l'ematoma dell'ileo-psoas con compressione del nervo femorale che può determinare problemi trofici all'arto inferiore. Ci possono essere sanguinamenti cerebrali, agli avambracci, ematuria e tutta una serie di manifestazioni emorragiche (ecchimosi e sanguinamenti gengivali). Vi ricordo il segno classico dell'emartro con anchilosi deformanti. La presentazione clinica dipenderà dalla gravità della deficienza del fattore VIII. Se la deficienza è grave si avranno già dei problemi alla nascita (sanguinamento dal cordone ombelicale, sanguinamenti cerebrali). Si capisce che il bambino ha dei problemi quando inizia a gattonare perché a livello delle ginocchia ci saranno delle tumefazioni con emartri. Nelle forme moderate si riscontreranno sanguinamenti all'eruzione dei denti e se il bambino è ebreo durante la circoncisione. Nell’800 l’emofilia colpì molti membri delle famiglie reali di Inghilterra, Spagna, Germania e Russia. Venne definita la malattia dei re. Tutti i soggetti colpiti erano discendenti diretti della regina Vittoria, la prima portatrice nota di emofilia nella sua famiglia. Forse il più famoso tra i discendenti di Vittoria colpiti dall’emofilia è il figlio dello Zar Nicola II, Alessandro, sterminato poi con la sua famiglia durante la rivoluzione bolscevica del 1917. In molte foto lo Zar veniva fotografato con stratagemmi (tipo mettere il ginocchio sullo scalino) per nascondere l'anchilosi a livello del ginocchio. A seguito della malattia del bambino venne chiamato a corte Rasputin che ebbe influenze negative sullo Zar. Si formò poi la prima e la seconda Duma ed infine il bambino venne fucilato. La diagnosi si sospetta quando viene da voi un bambino con problemi di sanguinamento e PTT allungato. L'emofilia può anche essere acquisita per la presenza di anticorpi contro il fattore VIII o il fattore IX. Questo può succedere nelle malattie autoimmuni o nelle sindromi paraneoplastiche. Il trattamento per l'emofilia consiste nella terapia sostitutiva, cioè nella somministrazione del fattore mancante (fattore VIII nell'emofilia A, fattore IX nella B). Fino a pochi anni fa questa era una pratica necessaria ma rischiosa, perché l'unico modo per ottenere questi fattori era quello di concentrarli partendo dal sangue di molti donatori, con un elevato rischio di contrarre virus, in particolare l'HIV. Tra il '78 e l'85 migliaia di emofilici sono stati sterminati dall'HIV. In Italia in quel periodo abbiamo avuto meno morti perché da sempre da noi veniva seguita una terapia con desmopressina. La desmopressina, derivato della vasopressina, spreme il fattore VIII dall'endotelio ed aumenta così i livelli di fattore VIII, se pur di poco,permettendo di trattare i pazienti con emofilia moderata. Oggi invece le tecniche di ingegneria genetica permettono di ottenere gli stessi fattori (fattore VIII ricombinante) in grande quantità, senza la necessità di ricorrere a donatori, evitando così ogni pericolo di infezione. MALATTIA DI VON WILLEBRAND E' causata da una carenza quantitativa del fattore di Von Willebrand, necessario a far aderire le piastrine al subendotelio. Serve anche a veicolare il fattore VIII fungendo da carrier. E' dunque una patologia mista di natura piastrinica e coagulativa. Esistono diverse varianti della malattia ma la più importante è quella di tipo II. L a sintomatologia è varia, si avranno: • • • sanguinamento moderato, solitamente post-traumatico, sanguinamenti di cute e mucose (difetto piastrinico) emartri ed ematomi (nelle forme più gravi) Nella maggior parte dei casi non rappresenta una grave patologia. I sanguinamenti si verificano alla seconda-terza decade di vita più spesso a seguito di traumi abbastanza importanti. FATTORI VITAMINA K DIPENDENTI Sono i fattori II, VII ed i IX ed il X. Il mio trucco per ricordarli è quello di memorizzare la data 1972. Anche la proteina C e la proteina S sono vitamina K dipendenti. La vitamina K è prodotta dall'intestino da alcuni batteri. La vitamina K serve a carbossilare l'acido glutammico di queste proteine. In mancanza di vitamina K tali fattori non sono funzionanti. La deficienza di vitamina K è principalmente causata da deficit nutritivo e da alcuni antibiotici, che alterano la flora batterica necessaria alla sintesi della vitamina. Il difetto di VII altera subito il PT. Dunque il PT rappresenta il test migliore per studiare l'eventuale deficit di vitamina K. Per valutare se il deficit si corregge con somministrazione di vitamina K si fa sempre il PT. Tra le altre cause acquisite, oltre al deficit di vitamina K ed all'emofilia acquisita, di alterazione della coagulazione dobbiamo ricordare le malattie epatiche (il fegato produce i fattori della coagulazione) e la CID. La CID è una condizione molto grave. Per effetto di danno endoteliale, per eccessiva attivazione macrofagica e per rilascio di TF si verifica l'attivazione della coagulazione che provoca trombosi ma allo stesso tempo un consumo dei fattori della coagulazione con eventuali sanguinamenti. A tale processo consegue una reazione omeostatica di equilibrio di iperfibrinolisi secondaria. Le caratteristiche sono: • • • riduzione del numero delle piastrine e del fibrinogeno aumento del PT e PTT presenza di D-dimero e fibrinopeptidi, perché si verifica trombosi e fibrinolisi. La sepsi, le neoplasie (carcinomi e la leucemia acuta promielocitica), i traumi, gli interventi chirurgici, le epatopatie, le complicanze ostretiche (ritenzione di feto morto, l'aborto, la placenta previa), morso di alcuni serpenti sono condizioni in grado di determinare CID. Vi prego di porre attenzione a questa slide in cui vengono considerati gli esami di primo livello (BT, numero di piastrine, PT e PTT). Se abbiamo un disordine vascolare→ normali :PT, PTT e numero di piastrine allungato il tempo di sanguinamento Se abbiamo una anomalia piastrinica qualitativa si verificherà un allungamento del bleeding time. Se invece il deficit piastrinico è quantitativo si avrà pure la riduzione del numero di piastrine. In entrambi i casi PT e PTT saranno normali. Se abbiamo un deficit congenito del fattore VIII o del IX( emofilia) avremo un allungamento del PTT con gli altri parametri nella norma. In caso di deficit di vitamina K o epatopatia chiaramente avremo un allungamento del PT e del PTT. In caso di CID avremo allungamento sia del bleeding time (le piastrine si consumano durante la CID) sia del PT e del PTT per consumo dei fattori della coagulazione. Lezione 16 VALUTAZIONE DELL’EMOSTASI La volta scorsa si è parlato dell'emofilia, del Von Willebrand, della carenza dei fattori vitamina Kdipendenti , della CID coagulazione intravascolare disseminata. Prima di completare il discorso sull’emostasi ricordiamo a cosa servono gli esami si fanno di routine che sono il PTT e l'attività protrombinica (PT): → il PTT va a studiare la via intrinseca e quindi il fattore XII, il fattore XI, il fattore IX e il fattore VIII; → il tempo di protrombina o attività protrombinica studia soltanto il fattore VII; e poi ovviamente ambedue studiano la via comune. Il prof vuole ribadire che gli esami di primo livello che servono a valutare l'emostasi sono: il tempo di sanguinamento; la conta delle piastrine; l'attività protrombinica, il PTT. E sono rappresentate le varie alterazioni in maniera molto grossolana, cioè le anomalie vascolari determinano un'alterazione soltanto del tempo di sanguinamento, mentre tutto il resto è normale; le anomalie delle piastrine se le piastrine sono ridotte di numero ovviamente ci sarà una riduzione del numero di piastrine, ma sia se le piastrine sono ridotte di numero, sia se le piastrine sono mal funzionanti ambedue le condizioni provocano un allungamento del tempo di sanguinamento; se c’è un difetto congenito del fattore VIII o del fattore IX, fondamentalmente le emofilie, in questo caso non avremo un allungamento del tempo di sanguinamento, non avremo una piastrinopenia, non avremo una alterazione dell’attività protrombinica, ma avremo un allungamento del PTT; se abbiamo un paziente con malattia epatica o un paziente con un difetto dei fattori vitamina K correlati, in questo caso il tempo di sanguinamento sarà normale, le piastrine saranno normali, ma ci sarà un allungamento dell’attività protrombinica e del PTT, più spiccata a carico dell’attività protrombinica; infine se abbiamo un paziente con una coagulopatia da consumo avremo un tempo di sanguinamento allungato, una ridotte conta delle piastrine e un allungamento sia del PTT che del PT. PATOLOGIE DELL’ ENDOTELIO Parliamo di un’altra patologia che interessa appunto l’endotelio. L’endotelio è una macchina molto complessa che è in qualche modo refrattaria all’aggregazione piastrinica: ci sono tutta una serie di sostanze che vengono prodotte dall’endotelio e vengono secrete dall’endotelio con attività sia anticoagulante che procoagulante, ed esiste un equilibrio fra queste sostanze e fondamentalmente l’endotelio ha un meccanismo antitrombotico in condizioni normali: esistono meccanismi anticoagulanti, meccanismi antiaggreganti e anche meccanismi fibrinolitici. Tutto questo per dire che ci sono delle patologie dell’endotelio che possono portare ad una grave condizione. 1 Porpora Trombotica Trombocitopenica In particolare questo (slide) è uno striscio di sangue periferico di un paziente affetto da Porpora Trombotica Trombocitopenica o Sindrome di Moschowitz, ne abbiamo già accennato quando abbiamo parlato delle anemie, perché questa condizione è una anemia emolitica microangiopatica, cioè c’è una alterazione dei piccoli vasi che provoca la formazione di coaguli di fibrina, e questi coaguli di fibrina nei piccoli vasi non fanno altro che rompere i globuli rossi che vi passano attraverso, e quindi alla fine nel sangue periferico si osservano dei frammenti di globuli rossi, dei piccoli frammenti, dei globuli rossi un po’ “morsicati”, che sono i cosiddetti schistociti o schizociti che sono patognomonici di una anemia emolitica microangiopatica, cioè da distruzione di globuli rossi legata al fatto che questi globuli rossi, passando attraverso questi coaguli di fibrina si rompono. Questa condizione (si vedono altri elementi in questo modo, o elementi cosiddetti a elmetto, che sono appunto schistociti) è tipica delle anemie emolitiche microangiopatiche delle quali fanno parte la CID, coagulazione intravascolare disseminata, l'abbiamo già vista, nella CID si formano questi coaguli all'interno dei microvasi e ci possono essere questi schizociti; le altre due condizioni sono la porpora trombotica trombocitopenica e la sindrome uremico emolitica, che si differenziano per il fatto che nella sindrome uremico emolitica questo processo avviene soprattutto a carico dei vasi renali, mentre invece nella porpora trombotica trombocitopenica è un processo generalizzato, con maggior interessamento anzi dei vasi cerebrali rispetto ad altri vasi. FISIOPATOLOGIA Qual è la fisiopatologia di queste condizioni? Il fattore di Von Willebrand serve a legare le piastrine fra di loro e a farle aderire al subendotelio, permette l'aggregazione piastrinica e l'adesione piastrinica al sub endotelio. In condizioni normali il fattore di Von Willebrand viene prodotto dalle cellule endoteliali, le quali producono dei grossi multimeri di questo fattore, il quale poi viene scisso da una protesi per cui questi multimeri diventano molto più piccoli e vanno a fare il loro lavoro cercando appunto di fare aderire le piastrine. Questa proteasi si chiama ADAMTS 13, da questo acronimo che significa a disintegrin and metalloproteinase with a thrombospondin. Quindi in condizioni normali l’ADAMTS 13 non fa altro che scindere questi grossi multimeri di fattore di Von Willebrand che sono secreti dalla cellula endoteliale: (slide) questa è la cellula endoteliale, questa è la sua membrana, a questo livello sta attaccato l’adamts 13 e appena la cellula endoteliale produce questo grosso multimero di fattore di Von Willebrand l’adamts13 subito lo cliva e produce dei multimeri molto più piccoli. In questi pazienti con PTT (porpora trombotica trombocitopenica), succede che manca l’ADAMTS 13 e quindi questi grossi multimeri che vengono prodotto hanno una maggiore attività aggregante le piastrine rispetto ai multimeri di minori dimensioni, tutto questo provoca appunto una aggregazione piastrinica che è responsabile poi della formazione dei trombi di fibrina e responsabile poi di tutta la sintomatologia di questi pazienti. Questa liberazione di grossi multimeri di fattore di Von Willebrand può essere anche stimolata da alcune citochine ma soprattutto viene stimolata dalla tossina della Shigella, per cui alcune infezioni intestinali aumentano la liberazione di questi grossi multimeri. In questo caso la Shigella determina questa alterazione a carico delle cellule endoteliali dei vasi renali per cui con lo stesso meccanismo la tossina della shigella provoca a livello di queste cellule endoteliali la liberazione di questo grosso multimero di fattore di Von Willebrand che va a fare aderire le piastrine e quindi si forma il trombo piastrinico. Quello che manca è appunto questa Adamts 13, questa proteasi ed è stato dimostrato che i pazienti affetti da PTT durante gli episodi acuti hanno un livello molto basso di questa proteasi, che invece migliora col trattamento e quando i pazienti sono in remissione. 2 CLINICA E’ importante riconoscere questa malattia, come si è detto a proposito delle anemie, perché questi sono pazienti che presentano una tipica espressione clinica: sono pazienti anemici perché hanno questa anemia emolitica microangiopatica, quindi hanno una anemia in cui ci sono le caratteristiche laboratoristiche dell’emolisi, quindi ci sarà la bilirubina aumentata (bilirubina indiretta), ci sarà l’LDH aumentato, ci sarà l’aptoglobina bassa, ci saranno i reticolociti aumentati. Questi pazienti sono anche piastrinopenici perché c’è appunto un consumo di piastrine legato al meccanismo della liberazione del fattore di Von Willebrand, quindi l’altro cardine diagnostico è la piastrinopenia; sono pazienti che hanno una insufficienza renale perché questa microangiopatia interessa i reni comunque, anche se la PTT interessa anche il cervello e per questo motivo hanno dei disturbi neurologici che sono dei disturbi quanto mai vari, possono andare dalla semplice cefalea al coma, oppure anche disturbi psichiatrici; e infine hanno la febbre: quindi questa è la pentade sintomatologica 1. 2. 3. 4. 5. anemia emolitica microangiopatica piastrinopenia insufficienza renale disturbi neurologici febbre. Questi pazienti se non si fa la diagnosi muoiono a causa di questa compromissione cerebrale; se si fa la diagnosi basta fare la plasmaferesi (plasma-exchange), cioè si toglie il plasma del paziente che è carente di Adamts 13 e ci si mette un plasma normale in cui è presente l’Adamts 13. (slide) Questa è la curva di sopravvivenza di questi pazienti che hanno fatto una plasmaferesi adeguata con 22 l di plasma, sommando le varie plasmaferesi, con una plasmaferesi al giorno per una settimana e poi dopo si allungano gli intervalli, uno ogni 2 giorni, ogni 3 giorni... Quindi è importante fare la diagnosi perché si passa da una mortalità dall’80% ad una guarigione dell’80%, quindi ricordiamo che se il paziente si presente con una piastrinopenia, disturbi neurologici, una anemia emolitica microangiopatica, se poi il paziente ha pure la febbre, ha una insufficienza renale e ha pure gli schistociti osservati allo striscio di sangue periferico questi pazienti hanno la sindrome di Moschowitz o PTT. TROMBOSI La diagnosi clinica di una tromboflebite è molto semplice: si vede un arto inferiore che ha una grandezza doppia rispetto l’arto non colpito dalla trombosi, è arrossato è caldo ecc... presenta tutti i segni della flogosi. Quali sono le cause di trombosi? Noi possiamo avere trombosi venose e trombosi arteriose. Ma per quanto riguarda le cause, la famosa triade di Virchoff è rappresentata da: le modifiche nello scorrimento del sangue; le modifiche nella composizione del sangue; l’alterazione della parete vasale. Quindi un rallentamento dello scorrimento del sangue, Una modifica della composizione, cosa vuol dire alterazione della composizione del sangue? 3 Abbiamo parlato della policitemia vera: qui c’è un aumento del numero dei globuli rossi con alterazione della composizione del sangue; e ovviamente tutte le alterazione dei vasi che innescano un processo trombotico e che hanno un significato più elevato quando si parla di trombosi arteriosa: tipicamente le trombosi delle arterie coronariche sono trombosi che si innescano perché lì c’è la placca ateromasica che sicuramente costituisce una alterazione della parete vascolare. Quindi i fattori legati alle trombosi sono: la stasi, intesa come una condizione di incremento dell’età e incremento dell’immobilizzazione, condizioni che tra l’altro vanno di pari passo, quindi il paziente anziano che si muove molto meno è sicuramente un paziente che ha un problema di immobilizzazione, ma anche immobilizzazione forzata in un paziente giovane, per esempio uno che si rompe una gamba sta a letto 120 giorni, quella è una immobilizzazione forzata; poi ci possono essere delle condizioni in cui ci sono dei difetti degli anticoagulanti: abbiamo visto prima che l’emostasi è una vera e propria bilancia, ci sono fattori anticoagulanti e fattori procoagulanti, i fattori procoagulanti sono tutte quelle proteasi, fattore II, fattore X... che hanno un effetto procoagulante, le sostanze anticoagulanti sono l’antitrombina III, la proteina C, la proteina S e poi ci sono tutta una serie di altre sostanze anticoagulanti; quindi ci possono essere dei difetti degli anticoagulanti, tipico il difetto di antitrombina III, oppure un eccesso di procoagulanti, oppure anche alcuni ormoni favoriscono l’ eccesso di coagulazione, ormoni quali per esempio gli estrogeni, come sappiamo la pillola anticoncezionale è un forte agente procoagulante; e poi nelle neoplasie si liberano tante citochine cha hanno un effetto procoagulante. Quindi tutte queste condizioni possono favorire la formazione di trombosi venose profonde, delle condizioni abbastanza rare che però possono complicarsi in un’ embolia polmonare, e questo è il problema grosso delle TVP, perché se il trombo si stacca e parte l’embolo, questo va a finire nel polmone e provoca l’embolia polmonare e l’embolia polmonare è una condizione che può essere mortale. Sintomatologia Inoltre pazienti che hanno delle TVP soprattutto recidivanti possono avere una condizione post trombotica con una discolorazione, un gonfiore e un senso di disturbo a livello delle gambe, tipicamente le persone anziane, soprattutto le donne che hanno fatto molti figli, perché la gravidanza è una condizione protrombotica. Ma ci possono essere anche dolori e ulcerazioni sempre a causa di questi disturbi trofici. Fattori di rischio Chiaramente è una condizione che aumenta con l’età e ci sono alcuni fattori di rischio genetici quali appunto il deficit di proteina C, il deficit di proteina S, il difetto della antitrombina, soprattutto della antitrombina III, e poi il cosiddetto fattore V Leyden e la mutazione del fattore II. Questi, il fattore V e il fattore II alterato, in pratica sono delle mutazioni dei geni che codificano per questi fattori che rendono i due fattori insensibili all’azione degli anticoagulanti, perché normalmente l’anticoagulante agisce inattivando alcuni fattori della coagulazione, fra questi appunto il fattore V e il fattore II: se questi due fattori sono mutati e quindi non sono più sensibili agli anticoagulanti, la bilancia si sposta in senso procoagulante. I contraccettivi orali sono dei potenti farmaci procoagulanti e questo per esempio è stata la causa della morte di questa ragazza di 28 anni che è morta per embolia polmonare subito dopo che era arrivata all’aeroporto di Heathrow dopo un viaggio di 20 ore proveniente dall’Australia. Da qui è nata la Sindrome della classe economica, è proprio il fatto che i pazienti che stanno per lungo tempo immobili, questa ragazza è stata 20 ore in classe economica, quindi in uno spazio stretto, immobile perché non si poteva muovere, e aveva sicuramente una diatesi trombotica, 4 si è fatta la TVP, appena si è mobilizzata è partito un embolo dal trombo che è andato a finire ai polmoni e questa ragazza è morta in aeroporto. Quindi quali sono i fattori di rischio della TVP? Possono essere fattori acquisiti e fattori ereditari; possono essere fattori secondari alla stasi, in pratica tutti fattori acquisiti, quali: l’immobilizzazione dopo un intervento chirurgico per esempio, l’obesità, l’insufficienza cardiaca, stroke ecc., la gravidanza, è un fattore acquisito che favorisce la trombosi e questo è normale perché quando il bambino nasce l’organismo si deve difendere dall’emorragia post partum, quindi la bilancia emostatica si sposta in senso protrombotico e più ci si avvicina al parto più aumenta il rischio di fatti trombotici. Condizioni congenite legate appunto a difetti ereditari dell’emostasi sono: il difetto della antitrombina III, difetto di proteina C, difetto di proteina S, oppure la resistenza del fattore V per esempio o del fattore II, ma ce ne sono tante altre, per esempio l’omocisteinemia, è un’altra condizione che può favorire la trombosi anche se ha un peso di gran lunga inferiore rispetto agli altri difetti. Fra le condizioni che alterano il sangue ci possono essere tutte quelle condizioni in cui c’è una ispissatio sanguinis, per esempio per fatti traumatici oppure per esempio interventi chirurgici, le neoplasie, i contraccetivi orali ecc. Poi ci possono essere le condizioni legate a disordini piastrinici: l’aumento delle piastrine, tutte le malattie mieloproliferative croniche provocano una alterazione non solo numerica ma anche funzionale delle piastrine. Poi importante è l’età; tutte le condizioni di sepsi possono associarsi a fatti trombotici. Diagnosi Allora proprio perché esiste un numero molto elevato di difetti genetici che possono favorire trombosi c’è purtroppo una corsa a fare tutti i test genetici che possono avere in qualche modo un interesse nel coinvolgimento dei fatti trombotici. Si vedono pazienti che arrivano con sfilze lunghissime di esami che servono solo a fare arricchire i laboratori ma che spesso non hanno un peso clinico tale da giustificare alcun trattamento, per cui sono ad es. pazienti eterozigoti per il gene della tetraidrofolatoriduttasi, che non hanno alcuna necessità di fare la terapia, ma che fanno questi test in maniera inutile: tutti questi test fanno fatti soltanto in pazienti che hanno trombosi venose, quando sono di età inferiore ai 40 anni, quando ci sono episodi ricorrenti, quando c’è assenza di fattori favorenti, una storia familiare positiva e trombosi venose in sedi insolite, prima fra tutti il sistema portale: in questo caso vanno eseguiti test di screening per le trombofilie eredofamiliari; e poi in alcune condizioni molto rare tipo l’ipertensione polmonare essenziale, trombosi arteriose in pazienti di età inferiore ai 40 anni. Fondamentalmente ci devono essere tutte queste condizioni perché vengano giustificati tutti questi 5 test costosi spesso fatti in maniera inutile. Uno dei Fattori più importanti che determina rischio di trombosi è la neoplasia: ci sono numerosi studi che dimostrano come pazienti neoplastici hanno maggior rischio di trombosi, e anche pazienti con mieloma che vengono trattati con talidomide hanno un maggior rischio di trombosi per cui in questi pazienti deve essere somministrata una profilassi antitrombotica. Un fattore che interviene molto nella genesi delle trombosi è il fatto che spesso i pazienti affetti da neoplasie, a questi viene inserito un catetere venoso centrale che serve a fare le frequenti infusioni di farmaci antiblastici e il catetere venoso centrale è il fattore più importante nello sviluppo di eventi trombotici: fino al 20% di eventi trombotici in pazienti affetti da malattie ematologiche maligne che presentavano un catetere venoso centrale. Pertanto in presenza di un catetere venoso centrale il rischio di sviluppare fatti trombotici anche in pazienti che hanno malattie ematologiche, la maggior parte di questi pazienti spesso viaggiano con valori molto bassi di piastrine, per via della chemioterapia che fanno, perché il midollo spesso è interessato, ma nonostante questo il rischio di sviluppare degli eventi trombotici è molto elevato. 6
© Copyright 2026 Paperzz