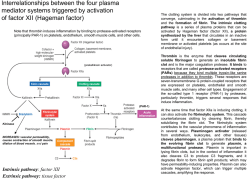
Interrelationships between the four plasma mediator systems triggered by activation of factor XII (Hageman factor) Note that thrombin induces inflammation by binding to protease-activated receptors (principally PAR-1) on platelets, endothelium, smooth muscle cells, and other cells. The clotting system is divided into two pathways that converge, culminating in the activation of thrombin and the formation of fibrin. The intrinsic clotting pathway is a series of plasma proteins that can be activated by Hageman factor (factor XII), a protein synthesized by the liver that circulates in an inactive form until it encounters collagen or basement membrane or activated platelets (as occurs at the site of endothelial injury). Thrombin is the enzyme that cleaves circulating soluble fibrinogen to generate an insoluble fibrin clot and is the major coagulation protease. It binds to receptors that are called protease-activated receptors (PARs) because they bind multiple trypsin-like serine proteases in addition to thrombin. These receptors are seven-transmembrane G protein-coupled receptors that are expressed on platelets, endothelial and smooth muscle cells, and many other cell types. Engagement of the so-called type 1 receptor (PAR-1) by proteases, particularly thrombin, triggers several responses that induce inflammation. (prekallikrein activator) (PAR-1) Kininase INCREASES: vascular permeability, causes contraction of smooth muscle, dilation of blood vessels, and pain Intrinsic pathway: factor XII Extrinsic pathway: tissue factor At the same time that factor XIIa is inducing clotting, it can also activate the fibrinolytic system. This cascade counterbalances clotting by cleaving fibrin, thereby solubilizing the fibrin clot. The fibrinolytic system contributes to the vascular phenomena of inflammation in several ways. Plasminogen activator (released from endothelium, leukocytes, and other tissues) cleaves plasminogen, a plasma protein that binds to the evolving fibrin clot to generate plasmin, a multifunctional protease. Plasmin is important in lysing fibrin clots, but in the context of inflammation it also cleaves C3 to produce C3 fragments, and it degrades fibrin to form fibrin split products, which may have permeability-inducing properties. Plasmin can also activate Hageman factor, which can trigger multiple cascades, amplifying the response. L’emostasi può essere vista come una successione di quattro eventi separati ma correlati • • • • fase parietale, fase endotelio-piastrinica; fase plasmatica o della coagulazione; fase trombo-dinamica. Fase vascolare • Vasocostrizione (La contrazione attiva delle fibre muscolari circolari lisce in corrispondenza o a monte della lesione di continuo è dovuta inizialmente ad un riflesso vasomotorio neurovegetativo e successivamente all’azione di sostanze liberate dalle piastrine: serotonina, catecolamine e TXA2 (dotato di spiccata attività aggregante piastrinica e vasocostrittiva). La vasocostrizione riduce transitoriamente l’intensità dell’emorragia e favorisce la marginazione delle piastrine). • Risposta diretta delle cellule muscolari in risposta al trauma • Riflesso neurovegetativo • Liberazione locale di sostanze vasocostrittrici (endotelina, serotonina) • La lesione delle cellule endoteliali espone il tessuto connettivo sottoendoteliale altamente trombogenico, al quale le piastrine aderiscono con cambiamento nella forma piastrinica e una reazione di esocitosi/degranulazione. I fattori che si liberano dai granuli piastrinici (ADP, TXA2, serotonina ed altri) reclutano ulteriori piastrine che aggregano sopra le prime, così da formare il tappo piastrinico. Tale reazione piastrinica avviene entro pochi minuti dalla lesione e, insieme alla vasocostrizione, costituisce la cosidetta emostasi primaria Differentiation of hematopoietic cells SCF, stem cell factor; Flt3L, Flt3 ligand; GM-CSF, granulocyte-macrophage colony-stimulating factor; M-CSF, macrophage colony-stimulating factor; G-CSF, granulocyte colony-stimulating factor. (mpl: recettore per la trombopoietina) Fase endotelio-piastrinica Adesione, aggregazione e attivazione delle piastrine Allorché un vaso sanguigno subisce una lesione di continuo, il collageno della parete vasale viene a contatto con il sangue che l’attraversa: le piastrine aderiscono immediatamente alle fibrille del collageno esposto nella breccia e sono stimolate a contrarsi. Successivamente, altre piastrine presenti nel flusso sanguigno aderiscono al primo strato e nel tempo di circa un minuto viene a formarsi il cosiddetto tappo emostatico piastrinico o primario, che chiude la soluzione di continuo L’evento iniziale è l’adesione delle piastrine al collageno Le piastrine quando aderiscono al collageno cambiano forma, si distendono su di esso e perdono i granuli. A questo punto altre piastrine presenti nel sangue che scorre aderiscono a quelle che si sono già legate al collageno (conglutinazione), formando un aggregato di piastrine. L’aggregato di piastrine costituisce un tampone che impedisce l’ulteriore perdita di sangue ed è propriamente un trombo bianco. 1.Trombo Rosso: caratteristico delle vene, è costituito da emazie inglobate in abbondante fibrina 2.Trombo Bianco: caratteristico delle arterie, costituito da piastrine con poca fibrina 3.Trombo Misto: caratteristico del microcircolo, (vasi ad intermedia velocità di flusso), costituito da piastrine, emazie e fibrina. Piastrine • • • • • • Cellule discoidi anucleate – Diametro 2-3 µm – Vita media 9-12 gg discoide → sfera spinosa Livello normale: 150 000 – 400 000 /nl – Piastrinopoiesi stimolata da trombopoietina, interleukina 11 Contengono actina Membrana ricca di recettori (integrine) per: – Collagene – Fibronectina – Fibrinogeno – Laminina – Von Willebrand – Trombospondina Granuli: – Lisosomi – Corpi densi o granuli δ (contengono calcio, serotonina (aggreg piastrinica), pirofosfato, adenosin-difosfato e adenosin-trifosfato ) (stimolati da ADP, ATP, Ca++, serotonina) – Granuli α (contengono differenti proteine quali il fibrinogeno, trombospondina (si lega a fibrinogeno e favorisce aggregazione piastrinica), il fattore di crescita di derivazione piastrinica, il fattore di von Willebrand, il fattore V, la fibronectina, la β-tromboglobulina e il fattore neutralizzante l'eparina (fattore piastrinico 4). ) (stimolati da adesione e alcuni fattori della coagulazione) Attivatori: – ADP, epinefrina, collageno, trombina, PAF (platelet activating factor), complessi Ab-Ag, shear stress Fase piastrinica • • • • La lesione espone il sottoendotelio (collagene, proteoglicani, fibronectina) Adesione delle piastrine tramite integrine – Stabilizzazione dell’adesione tramite fattore di von Willebrand (vWF) Cambiamento conformazione discoide → sfera spinosa , dovuto a: – Polimerizzazione ATP-dipendente dell’actina – Aumento di Ca++ e liberazione del contenuto dei granuli (ADP, serotonina, fibrinogeno, trombina) – Esternalizzazione di fosfatidilserina (PF3) – Attivazione di fosfolipasi C (IP3→Ca++) e fosfolipasi A2 (→trombossano → aggregazione piastrinica) Aggregazione delle piastrine – Maggiormente stimolata da trombossano – Esposizione di recettori per fibrinogeno e formazione di ponti La fase di retrazione del coagulo è caratterizzata dalla cessione di acqua da parte del polimero di fibrina con il conseguente accorciamento dello stesso. Questa fase richiede un dispendio di energia sotto forma di ATP che viene prodotta dalle piastrine stesse ed è denominata metamorfosi viscosa. FP3 Attività coagulante delle piastrine piu’ nota è definita come fattore piastrinico 3 (FP3) (fosfolipide di membrana). E’ associata a fosfolipidi di membrana caricati negativamente che normalmente sono segregati sul versante interno della membrana piastrinica, ma vengono esposti alla superficie piastrinica allorchè queste vengono attivate. Il PF3 forma con i Ca++, il F IX ed il F VIII un complesso capace di legare ed attivare il F Xe quindi con i Ca++, il F V ed il F Xa un secondo complesso che lega la protrombina trasformandola in trombina. Structure and function of factor VIII-von Willebrand factor (vWF) complex Factor VIII is synthesized in the liver and kidney, and vWF is made in endothelial cells and megakaryocytes. The two associate to form a complex in the circulation. vWF is also present in the subendothelial matrix of normal blood vessels and the alpha granules of platelets. Following endothelial injury, exposure of subendothelial vWF causes adhesion of platelets, primarily via glycoprotein lb platelet receptor. Circulating vWF and vWF released from the alpha granules of activated platelets can bind exposed subendothelial matrix, further contributing to platelet adhesion and activation. Activated platelets form hemostatic aggregates; fibrinogen (and possibly vWF) participate in aggregation through bridging interactions with the platelet receptor GpIIb/III. Factor VIII takes part in the coagulation cascade as a cofactor in the activation of factor X on the surface of activated platelets. VIA ESTRINSECA VIA INTRINSECA Nella via estrinseca abbiamo la formazione del complesso TF-VIIa-Ca++: il Fattore Tissutale, liberato a livello della lesione, funge da recettore per il FVIIa ed in presenza del Ca++ forma un “complesso” attivando due reazioni: Trasformazione del FIX in FIXa, che si complessa con il FVIIIa – che viene dalla via intrinseca – in presenza di fosfolipidi e Ca++. Il fattore VIII non viene sintetizzato solo dal fegato, ma anche a vari altri livelli. Trasformazione del FX in FXa che si complessa con il FVa, con i fosfolipidi e con il Ca++ (via comune). Fattore Va è presente anche a livello delle piastrine; è una pro-accelerina, che viene espressa nella membrana piastrinica. Ha inizio quando il sangue viene a contatto con superfici cariche negativamente (collagene subendoteliale per es.). Partecipano alla sua determinazione i Fattori: XII Hageman, XI Christmas, IX, VIII, fino all’attivazione del fattore X. La lesione espone il sottoendotelio (collagene, proteoglicani, fibronectina) FP3) Tissue Factor (TF) ed altrimenti indicato come fattore III o CD142 FP3: fattore piastrinico 3 VIA COMUNE Trasformazione del FX in FXa che si complessa con il FVa, con i fosfolipidi e con il Ca++. Il complesso FXa-FVafosfolipidi-Ca++ è anche detto protrombinasi, perché attiva la protrombina in trombina. La trombina (serinoproteasi) trasforma il fibrinogeno in fibrina solubile. La trombina agisce anche sul fattore XIII attivandolo a FXIIIa, che converte la fibrina solubile in fibrina insolubile. ACTION METABOLITE Vasoconstriction Thromboxane A2, leukotrienes C4, D4, E4 Vasodilation PGI2, PGE1, PGE2, PGD2 Increased vascular permeability Leukotrienes C4, D4, E4 Chemotaxis, leukocyte adhesion Leukotriene B4, HETE, lipoxins fattore piastrinico 3 (FP3) High Ratio: Thromboxane/Prostacicline trombi Fase trombo-dinamica L’aumento nel plasma del contenuto in plasmina rappresenta un meccanismo di difesa destinato a bilanciare il meccanismo di coagulazione. L’attivazione del plasminogeno richiede l’intervento di un attivatore tessutale del plasminogeno (t-PA) e un attivatore del plasminogeno di tipo urochinasico (u-PA), derivanti da precursori presenti nel sangue e nei tessuti (endotelio dei vasi, leucociti, epiteli, mesoteli e cellule delle membrane sinoviali). Attivatori esogeni del plasminogeno possono essere alcuni prodotti batterici, come la streptochinasi, la stafilochinasi, l’urochinasi e la saliva di alcuni pipistrelli ematofagi (vampiri). Con la dissoluzione e il riassorbimento del coagulo si avvia contemporaneamente il processo di riparazione della ferita, al termine del quale si ricostruisce lo strato di cellule endoteliali e la parete vasale riacquista la sua normale struttura. Emorragia -Esterna o interna -Interna: ematomi a) petecchie (1-2 mm) è una piccola manifestazione cutanea emorragica dovuta alla carenza di fattori intrinseci principali della coagulazione b) porpora ( >3mm) La porpora che si evidenzia a livello delle mucose gastriche e dell'apparato uro-genitale, o della cute, è dovuta a un'alterazione della permeabilità vascolare che permette la fuoriuscita di piccole quantità di globuli rossi a causa della diminuzione del numero delle piastrine. La porpora trombotica trombocitopenica (PTT) Il sistema immunitario si rivolge contro piastrine e globuli rossi con anemia emolitica da microangiopatia. L'anemia comporta coagulazione con eccessivo uso di piastrine. c) ecchimosi (comunemente chiamato livido, è un tipo di contusione generalmente causata da impatto, nella quale i capillari sottocutanei sono danneggiati, permettendo al sangue di diffondersi nei tessuti adiacenti. rosso blu perche’ i GR vengono fagogitati dai macrofagi, poi blu verde perche’ l’emoglobina e’ covertita a bilirubina e poi giallo marrone perche’ l’emoglobina diventa emosiderina); d) grossi accumuli di sangue in alcune cavita’: -emotorace-sangue nella pleura, -emopericardio-sangue nel sacco pericardico etc..) Diatesi emorragiche: si identifica in un insieme di difetti “”minori”" del sistema emostatico, quali la parziale carenza di uno o più fattori, la insufficiente produzione di fibrinogeno, i difetti di degranulazione delle piastrine • Aumentata fragilità dei vasi (es.: scorbuto) • Carenza o disfunzione delle piastrine • Alterazione della coagulazione • Combinazione di queste cause Test: Disordini emorragici -Tempo di sanguinamento: misura tempo in cui un’incisione (ferita) smette di sanguinare. Valutazione in vivo della funzionalità piastrinica -Conta piastrinica: su campione di sangue trattato con anticoagulanti (con contaglobuli) -Tempo di protrombina: test che valuta la via intrinseca (fattore di Hageman mediata) e quella comune della coagulazione -Tempo di protrombina parziale: Test che valuta la via estrinseca (tissue factor-mediata) e comune della coagulazione -problemi vascolari (es Ehlers Danlos (difetto sintesi collagene), telangiectasia emorragica ereditaria (displasia di capillari e venule: la dilatazione patologica e l'aumento di numero dei capillari) -infezioni, farmaci) -piastrine: numero ridotto o disfunzionali (Glanzmann:sindrome emorragica autosomica recessiva, che colpisce la linea megacariocitaria ed è caratterizzata dalla perdita dell'aggregazione piastrinica. Il difetto molecolare consiste in una anomalia quantitativa e/o qualitativa dell'integrina aIIbb3. Questo recettore media il legame delle proteine di adesione, che assicurano la formazione degli aggregati piastrinici e la formazione di trombi nei siti danneggiati dei vasi sanguigni. Bernard-Soulier-insieme di disturbi a carattere autosomico recessivo o co-dominante riguardante le piastrine, in particolare il recettore del fattore di von Willebrand della coagulazione, composto dalle glicoproteine gpIb, gpV e gpIX storage pool-comprende un eterogeneo gruppo di disordini caratterizzati da un deficit nel contenuto dei granuli piastrinici -difetto di coagulazione (emofilia A e B, malattia di von Willebrand): malattia ereditaria dovuta ad un deficit del fattore VIII (A) o IX (B) della coagulazione. Entrambi I geni sono su cromosoma X Malattia di Von Villebrand colpisce entrambi isessi Difetto di aggregazione piastrinica Difetto nei megacariociti Difetti coagulativi piastrino-dipendenti • • • Anomalie congenite – Sindrome di von Willebrand – Sindrome di Bernard-Soulier: deficit del recettore Anomalie acquisite – Trombocitopenia (basso livello di piastrine) – Distruzione delle piastrine circolanti: leucemia, sindromi mieloproliferative, eccesso di aspirina etc Farmaci antipiastrinici: – Aspirina (inibisce cicloossigenasi e riduce trombossano) – Effetto duraturo (piastrine non hanno nucleo e non possono rigenerare gli enzimi) – Dipiramidolo (inibisce effetto di ADP) • Rigetto iperacuto (minuti-ore) Reazione antigene-anticorpo a livello endotelio vascolare • Rigetto acuto (cellulare e umorale) (entro pochi giorni) Concorrono lesioni cellulari e umorali • Rigetto cronico (entro mesi - creatina nel siero: composto intermedio del metabolismo energetico sintetizzato dal fegato) MECCANISMI DEL RIGETTO DEGLI ALLOTRAPIANTI Nei diversi modelli sperimentali sia cellule T CD4+ che CD8+, che alloanticorpi specifici sono in grado di mediare il rigetto di un allotrapianto (cioè un trapianto tra due individui geneticamente diversi, ma appartenenti alla stessa specie); inoltre, il rigetto può essere inibito da anticorpi anti-CD4 o antiCD8. Questi diversi sistemi effettori causano il rigetto attraverso meccanismi differenti. 1. I CTL (Linfociti T Citotossici) alloreattivi, rappresentati principalmente da linfociti T con fenotipo CD8+, lisano direttamente le cellule endoteliali e parenchimali del trapianto. 2. I linfociti T helper alloreattivi possono reclutare ed attivare i macrofagi, dando inizio al danno del tessuto trapiantato attraverso un meccanismo di tipo DTH (ipersensibilità ritardata). 3. Gli alloanticorpi si legano all'endotelio ed attivano il sistema del complemento, danneggiando i vasi sanguigni del tessuto trapiantato. Ipersensibilità di tipo IV Rigetto d’organo allogenico Schematic representation of the events that lead to the destruction of histoincompatible grafts Graft APC + host antigens B and C Host APC + graft antigens host Rigetto umorale damage of the graft by a local delayed hypersensitivity reaction induce tissue damage by a local delayed hypersensitivity reaction A In the direct pathway, donor class I and class II antigens on antigen-presenting cells in the graft (along with B7 molecules, not shown) are recognized by CD8+ cytotoxic T cells and CD4+ helper T cells, respectively, of the host. CD4+ cells proliferate and produce cytokines that induce tissue damage by a local delayed hypersensitivity reaction and stimulate B cells and CD8+ T cells. CD8+ T cells responding to graft antigens differentiate into cytotoxic T lymphocytes that kill graft cells. In the indirect pathway, graft antigens are displayed by host APCs and activate CD4+ T cells, which damage the graft by a local delayed hypersensitivity reaction. The example shown is of a kidney allograft. Difetto di aggregazione piastrinica Difetto nei megacariociti Shock • Scompenso ipossico di molti organi dovuto a collasso cardiocircolatorio da: • 1)Difetto di volume ( es emorragico) • 2)Difetto di pompa ( es: cardiogenico) • 3) Settico ( vasodilatazione sistemica, coagulazione intravascolare disseminata..) Three major types of shock Type of shock Clinical example Cardiogenic Hypovolemic Septic Principal mechanisms Failure of myocardial pump owing to intrinsic myocardial damage, extrinsic pressure, or obstruction to outflow Myocardial infarction Ventricular rupture Arrhythmia Cardiac tamponade Pulmonary embolism Hemorrhage Fluid loss, e.g., vomiting, diarrhea, burns, or trauma Inadequate blood or plasma volume Overwhelming microbal infections Endotoxic shock Gram-positive spticemia Fungal sepsis Superantigens Peripheral vasodilation and pooling of blood; endothelial activation/injury; leukocyteinduced damage; disseminated intravascular coagulation; activation of cytokine cascades ostruzione parziale o totale della circolazione arteriosa polmonare Fasi dello shock 1) Non progressiva : la funzionalita’ degli organi principali ( cuore, cervello, rene) e’preservata. Vasocostrizione periferica, tachicardia, ritenzione di liquidi ( renina-angiotensina-aldosterone) 2) Progressiva. Meccanismi di compensazione come possibile causa di danno. Tachicardia se prolungata ed eccessiva riduce la gittata, la vasocostrizione porta a danno ischemico cellulare con formazione di acido lattico e vasodilatazione Il sangue va al microcircolo periferico 3) Recupero e’ impossibile. Necrosi nei diversi organi Endotossina Componente della membrana batterica fatta da un core tossico detto lipide A ricoperto da polisaccaridi Lega a proteine seriche e poi al CD14 sui leucociti e l’endotelio Agisce direttamente ma anche tramite induzione di citochine infiammatorie Il riconoscimento di LPS Cascata citochinica Citochine: mediatori polipeptidici del sistema immunitario non antigene specifici che fungono da segnale di comunicazione fra le cellule del sistema immunitario e fra queste e diversi organi e tessuti Danno diffuso dei capillari alveolari determinante grave insufficienza respiratoria con ipossiemia arteriosa refrattaria alla somministrazione di ossigeno glycoprotein lb platelet receptor platelet receptor GpIIb/III sequenza arginina-glicina-acido aspartico (RGD) del fibrinogeno Vena iliaca: trasporta il sangue povero di ossigeno dalla metà inferiore del corpo al cuore Fase trombo-dinamica L’aumento nel plasma del contenuto in plasmina rappresenta un meccanismo di difesa destinato a bilanciare il meccanismo di coagulazione. L’attivazione del plasminogeno richiede l’intervento di un attivatore tessutale del plasminogeno (t-PA) e un attivatore del plasminogeno di tipo urochinasico (u-PA), derivanti da precursori presenti nel sangue e nei tessuti (endotelio dei vasi, leucociti, epiteli, mesoteli e cellule delle membrane sinoviali). Attivatori esogeni del plasminogeno possono essere alcuni prodotti batterici, come la streptochinasi, la stafilochinasi, l’urochinasi e la saliva di alcuni pipistrelli ematofagi (vampiri). Con la dissoluzione e il riassorbimento del coagulo si avvia contemporaneamente il processo di riparazione della ferita, al termine del quale si ricostruisce lo strato di cellule endoteliali e la parete vasale riacquista la sua normale struttura. Difetto di aggregazione piastrinica Difetto nei megacariociti Structure and function of factor VIII-von Willebrand factor (vWF) complex Factor VIII is synthesized in the liver and kidney, and vWF is made in endothelial cells and megakaryocytes. The two associate to form a complex in the circulation. vWF is also present in the subendothelial matrix of normal blood vessels and the alpha granules of platelets. Following endothelial injury, exposure of subendothelial vWF causes adhesion of platelets, primarily via glycoprotein lb platelet receptor. Circulating vWF and vWF released from the alpha granules of activated platelets can bind exposed subendothelial matrix, further contributing to platelet adhesion and activation. Activated platelets form hemostatic aggregates; fibrinogen (and possibly vWF) participate in aggregation through bridging interactions with the platelet receptor GpIIb/III. Factor VIII takes part in the coagulation cascade as a cofactor in the activation of factor X on the surface of activated platelets. ACTION METABOLITE Vasoconstriction Thromboxane A2, leukotrienes C4, D4, E4 Vasodilation PGI2, PGE1, PGE2, PGD2 Increased vascular permeability Leukotrienes C4, D4, E4 Chemotaxis, leukocyte adhesion Leukotriene B4, HETE, lipoxins fattore piastrinico 3 (FP3) High Ratio: Thromboxane/Prostacicline trombi Disordini emorragici - Tempo di sanguinamento, numero delle piastrine tempo di protrombina e tempo di protrombina parziale -problemi vascolari ( es sindrome Ehlers Danlos con collagene mutato; telangiectasia emorragica ereditaria (displasia dei vasi); infezioni, farmaci) -piastrine : numero ridotto o disfunzionali ( Glanzmann, Bernard-Soulier, storage pool) -coagulazione ( emofilia A: carenza fattore VIII; emofilia B: carenza fattore IX; malattia di von Willebrand: ridotti livelli di fattore di von Willebrand) Generation of arachidonic acid metabolites (eicosanoids) and their roles in inflammation The molecular targets of action of some anti-inflammatory drugs are indicated by a red X. COX, cyclooxygenase; HETE, hydroxyeicosatetraenoic acid; HPETE, hydroperoxyeicosatetraenoic acid. LTB4 is a potent chemotactic agent and activator of neutrophil functional responses, such as aggregation and adhesion of leukocytes to venular endothelium, generation of oxygen free radicals, and release of lysosomal enzymes When cells are activated by diverse stimuli, their membrane lipids are rapidly remodeled to generate biologically active lipid mediators that serve as intracellular or extracellular signals to affect a variety of biologic processes, including inflammation and hemostasis. These lipid mediators are thought of as autocoids, or short-range hormones that are formed rapidly, exert their effects locally, and then either decay spontaneously or are destroyed enzymatically. activated by stress or various stimuli, as eg. C5a) cytoplasmic Ca2+ from dietary sources or by conversion from the essential fatty acid linoleic acid predominant enzyme in neutrophils COX-1 is responsible for the production of prostaglandins that are involved in inflammation but also serve a homeostatic function (e.g., fluid and electrolyte balance in the kidneys, cytoprotection in the gastrointestinal tract). In contrast, COX-2 stimulates the production of the prostaglandins that are involved in inflammatory reactions. A thromboxane-prostacyclin imbalance has been implicated as an early event in thrombus formation in coronary and cerebral blood vessels. The prostaglandins are also involved in the pathogenesis of pain and fever in inflammation. PGE2 is hyperalgesic in that it makes the skin hypersensitive to painful stimuli. The principal actions of lipoxins are to inhibit leukocyte recruitment and the cellular components of inflammation. ACTION METABOLITE Vasoconstriction Thromboxane A2, leukotrienes C4, D4, E4 Vasodilation PGI2, PGE1, PGE2, PGD2 Increased vascular permeability Leukotrienes C4, D4, E4 Chemotaxis, leukocyte adhesion Leukotriene B4, HETE, lipoxins ADH: ormone antidiuretico Causes of Thrombocytopenia Decreased production of platelets Generalized diseases of bone marrow Aplastic anemia: congenital and acquired Marrow infiltration: leukemia, disseminated cancer Selective impairment of platelet production Drug-induced: alcohol, thiazides, cytotoxic drugs Infections: measles, human immunodeficiency virus (HIV) Ineffective megakaryopoiesis Megaloblastic anemia Myelodysplastic syndromes Decreased platelet survival Immunologic destruction Autoimmune: idiopathic thrombocytopenic purpura, systemic lupus erythematosus Isoimmune: post-transfusion and neonatal Drug-associated: quinidine, heparin, sulfa compounds Infections: infectious mononucleosis, HIV infection, cytomegalovirus Nonimmunologic destruction Disseminated intravascular coagulation Thrombotic thrombocytopenic purpura Giant hemangiomas Microangiopathic hemolytic anemias Sequestration Hypersplenism Dilutional Major Disorders Associated with Disseminated Intravascular Coagulation Obstetric Complications Abruptio placentae Retained dead fetus Septic abortion Amniotic fluid embolism Toxemia Infections Gram-negative sepsis Meningococcemia Rocky Mountain spotted fever Histoplasmosis Aspergillosis Malaria Neoplasms Carcinomas of pancreas, prostate, lung, and stomach Acute promyelocytic leukemia Massive Tissue Injury Traumatic Burns Extensive surgery Miscellaneous Acute intravascular hemolysis, snakebite, giant hemangioma, shock, heat stroke, vasculitis, aortic aneurysm, liver disease Pathophysiology of disseminated intravascular coagulation Fase coagulativa VIA INTRINSECA Ha inizio quando il sangue viene a contatto con superfici cariche negativamente (collagene subendoteliale per es.). Partecipano alla sua determinazione i Fattori: XII Hageman, XI Christmas, IX, VIII, fino all’attivazione del fattore X. VIA ESTRINSECA Nella via estrinseca abbiamo la formazione del complesso TF-VIIa-Ca++: il Fattore Tissutale, liberato a livello della lesione, funge da recettore per il FVIIa ed in presenza del Ca++ forma un “complesso” attivando due reazioni: Trasformazione del FIX in FIXa, che si complessa con il FVIIIa – che viene dalla via intrinseca – in presenza di fosfolipidi e Ca++. Il fattore VIII non viene sintetizzato solo dal fegato, ma anche a vari altri livelli. Trasformazione del FX in FXa che si complessa con il FVa, con i fosfolipidi e con il Ca++ (via comune). Fattore Va è presente anche a livello delle piastrine; è una pro-accelerina, che viene espressa nella membrana piastrinica. VIA COMUNE Trasformazione del FX in FXa che si complessa con il FVa, con i fosfolipidi e con il Ca++ Il complesso FXa-FVa-fosfolipidi-Ca++ è anche detto protrombinasi, perché attiva la protrombina in trombina. La trombina (serinoproteasi) trasforma il fibrinogeno in fibrina solubile. La trombina agisce anche sul fattore XIII attivandolo a FXIIIa, che converte la fibrina solubile in fibrina insolubile. Cascata della coagulazione La lesione espone il sottoendotelio (collagene, proteoglicani, fibronectina) Tissue Factor (TF) ed altrimenti indicato come fattore III o CD142 Anemie •Funzioni globuli rossi è trasportare ossigeno ai tessuti periferici Diminuzione di tale capacità è solitamente causata da difetto nei globuli rossi o anemia (riduzione sotto i limiti normali della massa totale di globuli rossi corcolanti) •Riduzione rispetto al normale del volume dei globuli rossi sedimentato (ematocrito) •Riduzione della concentrazione di emoglobina nel sangue Classification of Anemia According to Underlying Mechanism Pathophysiologic Classification of Polycythemia Relative Reduced plasma volume (hemoconcentration) Es.: disidratazione per privazione d’H2O, vomito prolungato, diarrea, eccessivo uso di diuretici Absolute Primary Polycythemia vera, rare erythropoietin receptor mutations (low erythropoietin) Dovuta a alterazione intrinseca delle cellule staminali mieloid: è neoplasia di cellule mieloidii. Mutazioni del recettore dell’eritropoietina Secondary High erythropoietin (Spinge progenotori di globuli rossi verso aumentata eritropiesi) Appropriate: lung disease, high-altitude living, cyanotic heart disease Inappropriate: erythropoietin-secreting tumors (e.g., renal cell carcinoma, hepatocellular carcinoma, cerebellar hemangioblastoma) Disordini emorragici - Tempo di sanguinamento, numero delle piastrine tempo di protrombina e tempo di protrombina parziale -problemi vascolari ( es Ehlers Danlos, telangiectasia emorragica ereditaria, infezioni, farmaci) -piastrine : numero ridotto o disfunzionali ( Glanzmann, Bernard-Soulier, storage pool) -coagulazione ( emofilia A: carenza fattore VIII; e B: carenza fattore IX; Malattia di von Willebrand: ridotti livelli di fattore di von Willebrand) Generalità EMOSTASI L’alterazione dell’integrità del letto vascolare può dipendere da una lesione tissutale superficiale o da un grande trauma tissutale. Per ridurre il sanguinamento ed evitare la perdita di sangue dopo un danno tissutale sono attivati i componenti del sistema emostatico. I componenti di questo sistema dinamico integrato comprendono le piastrine, le cellule endoteliali ed i fattori plasmatici della coagulazione. Essi possono essere attivati o per l’esposizione nel letto vascolare a superfici estranee o da prodotti liberati dalle cellule danneggiate. L’emostasi può essere vista come una successione di quattro eventi separati ma correlati. Le principali fasi dell’emostasi possono essere riassunte schematicamente come segue: fase parietale, fase endotelio-piastrinica; fase plasmatica o della coagulazione; fase trombo-dinamica. Anche se per comodità descrittiva le diverse fasi vengono trattate separatamente esse in realtà avvengono pressoché contemporaneamente. Impossibile visualizzare l'immagine. Fase parietale Le cellule endoteliali formano una specie di rivestimento liscio e regolare della parete dei vasi sanguigni, che permette al sangue di scorrere regolarmente. Qualsiasi lesione della superficie interna di un vaso comporta l’interruzione di tale strato cellulare, per cui nella zona lesionata si verifica una iniziale vasocostrizione tendente a ridurre il calibro vasale. Questo evento contribuisce ad arrestare l’emorragia. La contrazione attiva delle fibre muscolari circolari lisce in corrispondenza o a monte della lesione di continuo è dovuta inizialmente ad un riflesso vasomotorio neurovegetativo e successivamente all’azione di sostanze liberate dalle piastrine: serotonina, catecolamine e TXA2. La vasocostrizione riduce transitoriamente l’intensità dell’emorragia e favorisce la marginazione delle piastrine. Nei vasi più piccoli (arteriole e venule) l’emostasi è devoluta quasi esclusivamente alla formazione di un tappo piastrinico, mentre nei capillari si potrebbe avere anche un semplice accollamento delle pareti favorito dalla formazione di una sottile pellicola di fibrina. Lesione vascolare e formazione del tappo emostatico primario. Fase endotelio-piastrinica Adesione, aggregazione e attivazione delle piastrine Allorché un vaso sanguigno subisce una lesione di continuo, il collageno della parete vasale viene a contatto con il sangue che l’attraversa: le piastrine aderiscono immediatamente alle fibrille del collageno esposto nella breccia e sono stimolate a contrarsi. Successivamente, altre piastrine presenti nel flusso sanguigno aderiscono al primo strato e nel tempo di circa un minuto viene a formarsi il cosiddetto tappo emostatico piastrinico o primario, che chiude la soluzione di continuo. L’evento iniziale è l’adesione delle piastrine al collageno Le piastrine quando aderiscono al collageno cambiano forma, si distendono su di esso e perdono i granuli. A questo punto altre piastrine presenti nel sangue che scorre aderiscono a quelle che si sono già legate al collageno (conglutinazione), formando un aggregato di piastrine. L’aggregato di piastrine costituisce un tampone che impedisce l’ulteriore perdita di sangue ed è propriamente un trombo bianco. Fase plasmatica o della coagulazione Il processo coagulativo può avvenire attraverso due sistemi: il sistema intrinseco; il sistema estrinseco. Il primo consiste nell’attivazione di fattori presenti nel plasma (da cui il termine intrinseco), mentre il secondo sistema si attiva quando al plasma viene aggiunto un estratto tessutale per cui, in contrapposizione al primo, si indica con il termine di estrinseco. In questa fase sono coinvolti prevalentemente i fattori della coagulazione ovvero proteine circolanti nel sangue e prodotte quasi tutte dal fegato. Se ne conoscono una dozzina circa, indicate in genere con un numero romano (es: fattore VII, VIII o IX) o con il nome proprio (es: fibrinogeno). Essi hanno la caratteristica peculiare di agire in sequenza, uno dietro l’altro, e ad ogni tappa il fattore che circola inattivo nel sangue, viene attivato ed agisce sul fattore successivo, che viene attivato a sua volta. Ad ogni tappa aumenta notevolmente il numero di molecole formate, così che alla fine di questa cascata coagulativa, partendo da poche molecole, si ottiene un numero enorme di molecole di fibrina. Per la produzione epatica di alcuni di questi fattori è essenziale la vitamina K. Sistema intrinseco I fattori indispensabili per tale sistema sono presenti nel plasma e solo una modificazione delle caratteristiche della parete vasale attiva le reazioni enzimatiche a cascata che conducono alla coagulazione. Il contatto del sangue con l’endotelio leso è indispensabile. Tale contatto con le fibre collagene e con il subendotelio rappresenta lo stimolo fisiologico per l’attivazione del fattore XII o fattore di Hageman. Il fattore XIIa attiva il fattore XI in fattore XIa, ma questa reazione procederebbe molto lentamente se il fattore XIIa, non trasformasse anche la precallicreina (fattore Fletchen) in callicreina. Il tutto verrebbe supportato dal chininogeno ad alto peso molecolare. Viene successivamente attivato il fattore IX o fattore Christmas in fattore IXa. Quest’ultimo in presenza di Ca++, fattore 3 piastrinico (PF3) e fattore VIII attiva il fattore X. Il fattore VII o fattore antiemofilico A è costituito da una grossa molecola glicoprotidica, composta da due differenti subunità, una ad alto e l’altra a basso peso molecolare. Nell’emofilia A è carente la porzione di piccole dimensioni del fattore VIII mentre la parte ad alto peso molecolare è presente. Essa si forma nelle cellule endoteliali e manca nella malattia di von Willebrand. Con l’attivazione del fattore X o fattore di Stuart-Prower originerà la via comune. Sistema estrinseco Con questo termine si intende il processo coagulativo che si attiva quando al plasma si aggiunge un estratto tissutale che normalmente è assente. La tromboplastina tessutale o fattore III si forma generalmente in seguito a necrosi dei tessuti. Essa insieme al Ca++ ed al fattore VII o proconvertina forma un complesso in grado di trasformare il fattore X in Xa (via comune). La formazione della fibrina attraverso questo sistema avviene nell’arco di tempo di 10-15 secondi, laddove il sistema intrinseco impiega 6-12 minuti. Pertanto la liberazione del fattore III attiva un meccanismo di difesa rapido nei confronti di un’ emorragia. Via comune Il fattore X attivato presenta una spiccata attività nel trasformare la protrombina o fattore II in trombina. Questa reazione è potenziata dalla presenza del fattore V proaccelerina del calcio e dei fosfolipidi piastrinici. Una volta formatasi la trombina, essa stacca proteoliticamente dal fibrinogeno o fattore I i fibrinpeptidi A e B e i monomeri di fibrina che successivamente polimerizzano spontaneamente tra di loro dando origine ad un gel di fibrina. Il reticolo di fibrina così formato si presenta scarsamente resistente. Per tale motivo il fattore XIII o fattore stabilizzante la fibrina interviene promuovendo la formazione di legami peptidici tra i monomeri di fibrina conferendo al coagulo una maggiore resistenza meccanica. Via Intrinseca e Via Estrinseca Fase trombo-dinamica Inizia pochi minuti dopo la formazione del coagulo e può essere divisa in due momenti: fase costruttiva fase distruttiva La fase costruttiva inizia con ispessimento e addensamento progressivo delle strutture fibrinose, seguito da riduzione del volume del coagulo (sineresi); successivamente, la fase costruttiva comporta la retrazione del coagulo, fenomeno al quale partecipa la trombostenina: la contrazione dei prolungamenti delle piastrine avvicina i filamenti di fibrina ai quali esse aderiscono tenacemente per il tramite della fibronectina con la fuoriuscita del siero. La retrazione del coagulo si completa dopo circa un’ora dall’inizio della sua formazione. La fase distruttiva subentra dopo alcuni giorni e comporta la dissoluzione del coagulo mediante fibrinolisi: la fibrina polimerizzata subisce l’idrolisi ad opera di un enzima, la plasmina o fibrinolisina, che si genera dal plasminogeno.
© Copyright 2026 Paperzz